
Whole-Exome Sequencing Identifies Pathogenic Variants in SMPD1, HIP1R, and POMC Associated with Obesity in a Consanguineous Pakistani Population
-
Shakeela Daud


Department of Biotechnology, Faculty of Life Sciences and Informatics, Balochistan University of Information Technology, Engineering and Management Sciences, Quetta, Pakistan
Sara NaudhaniDepartment of Biotechnology, Faculty of Life Sciences and Informatics, Balochistan University of Information Technology, Engineering and Management Sciences, Quetta, Pakistan
Sobia Faisal MalikDepartment of Environmental Science, Faculty of Life Sciences and Informatics, Balochistan University of Information Technology, Engineering and Management Sciences, Quetta, Pakistan
Hameeda PanezaiDepartment ofChemistry, Faculty of Basic Sciences, Balochistan University of Information Technology, Engineering and Management Sciences, Quetta, Pakistan
Fatima IqbalMultan Cancer Clinic, Nishtar Chowk, Nishtar Road, Multan, Pakistan
| Received 19 Nov, 2025 |
Accepted 14 May, 2026 |
Published 30 Jun, 2026 |
Background and Objective: Obesity, defined as a Body Mass Index (BMI) ≥30 kg/m2, is a multifactorial metabolic disorder arising from an imbalance between energy intake and expenditure. This study aimed to identify pathogenic variants in consanguineous Pakistani families with early-onset obesity to elucidate the genetic basis of the disorder. Materials and Methods: Twenty affected families and twenty sporadic obese individuals from various regions of Balochistan were recruited. Genomic DNA was extracted from peripheral blood, followed by whole-exome sequencing and validation using Sanger sequencing. Identified variants were analyzed using bioinformatics tools to predict their pathogenicity. Results: A heterozygous missense variant in the SMPD1 gene, segregating with autosomal dominant obesity, and previously reported variants in HIP1R and POMC genes associated with non-syndromic obesity phenotypes were identified. Conclusion: These findings broaden the mutational landscape of obesity-related genes, support genotype-phenotype correlations, and highlight the importance of genetic screening in consanguineous families for early diagnosis, counselling, and better understanding of hereditary obesity.
INTRODUCTION
Obesity is one of the most alarming global health problems and a major risk factor for metabolic disorders such as type II diabetes, cardiovascular diseases, and hypertension1. According to the World Health Organization, more than 1.9 billion adults worldwide are overweight, and over 650 million are obese2.
Obesity is defined as a body mass index (BMI) of 30 kg/m2 or greater and results from a chronic imbalance between energy intake and expenditure, leading to the excessive accumulation of triglycerides in adipose tissue3,4. While environmental factors, such as a sedentary lifestyle and high-calorie diets, contribute significantly, genetic predisposition also plays a critical role in determining susceptibility to obesity5. Heritability estimates suggest that more than 40% of BMI variation can be attributed to genetic factors6. Genetic studies have revealed that obesity is a heterogeneous disorder involving complex interactions between multiple genes and environmental factors. Among the known genetic contributors, monogenic forms of obesity caused by rare variants in single genes provide key insights into the mechanisms of energy homeostasis. The POMC (proopiomelanocortin) gene encodes a precursor protein crucial in appetite regulation through the leptin-melanocortin pathway; mutations in POMC are associated with early-onset severe obesity due to impaired satiety signaling7,8. The SMPD1 (sphingomyelin phosphodiesterase 1) gene encodes acid sphingomyelinase, an enzyme involved in sphingolipid metabolism; variants in SMPD1 have been linked to lipid storage abnormalities and syndromic forms of obesity9. Similarly, HIP1R (huntingtin-interacting protein 1-related) plays a role in endocytosis and cytoskeletal organization; alterations in HIP1R are reported to disrupt neuronal signaling pathways associated with non-syndromic obesity10. Despite advances in genomic technologies, the genetic landscape of obesity remains incompletely characterized, particularly in populations with high rates of consanguinity, such as Pakistan. The Pakistani population provides a unique opportunity to identify novel homozygous variants underlying monogenic and syndromic obesity due to its genetic structure11. Therefore, this study aimed to identify pathogenic variants in the SMPD1, HIP1R, and POMC genes among consanguineous families from Balochistan presenting with early-onset obesity. These findings contribute to expanding the mutational spectrum of obesity-related genes and improving understanding of genotype-phenotype correlations in underrepresented populations.
MATERIALS AND METHODS
Study area: This study was conducted in Quetta, Balochistan, Pakistan, during the year 2022-2023.
Study subjects and ethical approval: Twenty consanguineous families with early-onset obesity and twenty sporadic obese individuals were recruited from various regions of Balochistan, Pakistan. Written informed consent was obtained from all participants or their legal guardians before sample collection. The study protocol was approved by the Institutional Ethics Review Committee of BUITEMS, Quetta, and all procedures adhered to the ethical standards outlined in the Helsinki Declaration.
Clinical evaluation and sample collection: Detailed clinical and anthropometric data, including age, sex, height, weight, and Body Mass Index (BMI), were recorded for each participant. Peripheral blood samples (5 mL) were collected in EDTA-coated tubes from all affected individuals and available family members. Samples were stored at -20°C until further processing.
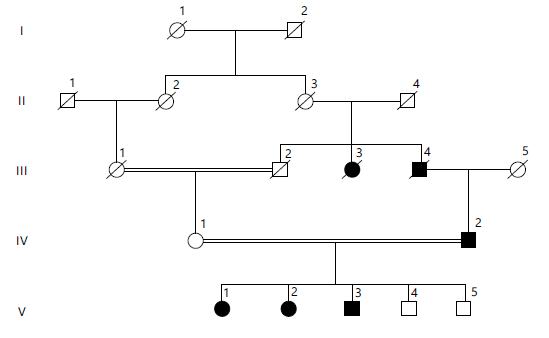
This family was sampled from the Luni District, Sibi, Balochistan. Peripheral blood samples were taken in Vacutainer tubes containing EDTA from all affected individuals, their parents, and normal siblings. Clinical evaluations and family histories were obtained through detailed interviews with all family members as shown in Fig. 1. This family is segregating with an obesity phenotype, with clinical features (comorbidities) involving type 2 diabetes, hypertension, CVD, and an eating disorder. All affected individuals showed similar, frequent clinical features that appeared at an early stage or developed during early childhood shown in Table 1.
| Table 1: | Clinical description of affected individuals in Family 4, including age, age of obesity onset, and Body Mass Index (BMI) with percentiles | |||
| Clinical Phenotype | Patient # → Clinical observation↓ | IV:2 | V:1 | V:2 | V:3 |
| Age | Age at the time of sampling | 37 | 10 | 8 | 7 |
| Obesity | Age of onset | 2.5 years | 1.5 year | 2 years | 1.8 year |
| BMI (kg/m2)/Percentile | 39.5 kg/m2 | 99th centile | 95th centile | 97th centile | |
| Age is reported in years. BMI percentiles are based on age. BMI: Body Mass Index (kg/m2). Roman numerals (IV, V) indicate generations, and Arabic numerals identify individuals within each generation (e.g., V:1 = first individual in the 5th generation) | |||||
|
|
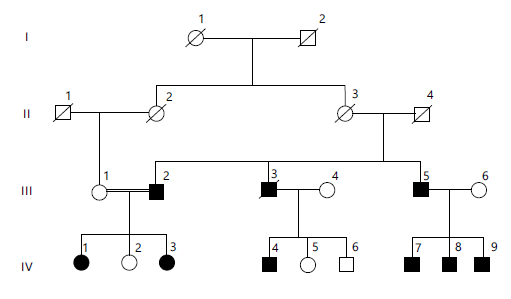
A diagonal line through a symbol indicates a deceased individual. Horizontal lines represent mating, and vertical lines indicate parent-offspring relationships. Roman numerals (I, II, III, IV, V) denote generations, and Arabic numerals identify individuals within each generation. Double horizontal lines indicate consanguineous marriages.
This family was sampled from Khajak District, Sibi, Balochistan. Clinical evaluation and family history were obtained by all members of the family as shown in Fig. 2. This family is segregated with an obesity phenotype, with clinical features involving type 2 diabetes, hypertension, CVD, and an eating disorder, inherited in an autosomal dominant manner (Table 2).
|
| Table 2: | Clinical description of the affected individuals of the family | |||
| Clinical phenotype |
Patient # → clinical observation↓ |
III:2 | III:5 | IV:1 | IV:3 | IV:4 | IV:7 | IV:8 | IV:9 |
| Age | Age at the time of sampling |
45 years | 32 years | 21 years | 20 years | 17 years | 5 years | 4 years | 2 years |
| Obesity | Age of onset BMI(kg/m2)/ |
2 years 40 kg/m2 |
2.5 years 39 kg/m2 |
3 years 42 kg/m2 |
2 years 35 kg/m2 |
2.5 years 98th centile |
1.8 year 96th centile |
2.5 years 95th centile |
1.5 year 97th centile |
| Type 2 diabetes |
Age of onset | 2.5 years | 3.5 years | 4 years | No | 3 years | 3 years | No | No |
| Hypertension | Yes/No | Yes | No | Yes | No | No | No | No | No |
| CVD | Yes/No | Yes | No | No | No | No | No | No | No |
| Eating disorder | Hyperphagia | Yes | No | Yes | No | Yes | Yes | Yes | Yes |
| Depression | Yes/No | Yes | Yes | Yes | Yes | Yes | No | No | No |
| BMI: Body Mass Index (kg/m2). Age is reported in years. A BMI percentiles are based on age- and sex-specific reference standards. “Yes/No” indicates the presence or absence of the clinical feature. Hyperphagia refers to excessive eating behavior. Roman numerals (III, IV) indicate generations, and Arabic numerals identify individuals within each generation (e.g., IV:1 = first individual in the 4th generation) | |||||||||
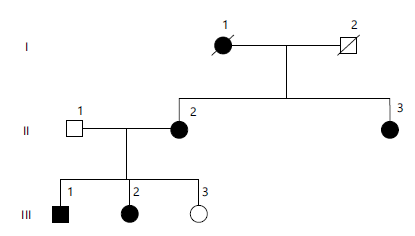
| Table 3: | Clinical description of the affected individuals of the family | |||
| Clinical Phenotype | Patient # →Clinical observation↓ | II:2 | III:1 | III:2 |
| Age | Age at the time of sampling | 36 | 13 | 12 |
| Obesity | Age of onset | 2 | 3 | 2.5 |
| BMI (kg/m2)/Percentile | 39 kg/m2 | 96th centile | 95th centile | |
| Type 2 diabetes | Age of onset | No | No | No |
| Hypertension | Yes/No | No | No | No |
| CVD | Yes/No | No | No | No |
| Eating disorder | Hyperphagia | Yes | Yes | Yes |
| Depression | Yes/No | Yes | Yes | No |
| Observations at birth | Delivery | Normal | Normal | Normal |
| BMI: Body Mass Index (kg/m2) and CVD: cardiovascular disease. Age is reported in years. A BMI percentiles are based on age- and sex-specific reference standards. “Yes/No” indicates the presence or absence of the clinical feature. Hyperphagia refers to excessive eating behavior | ||||
This family was ascertained from Jaffarabad, Balochistan. A non-consanguineous family as show in Fig. 3. This family exhibits a segregated obesity phenotype; all affected individuals display similar, frequent clinical features (comorbidities) that typically appear at an early stage or develop during early childhood, inherited in an autosomal dominant manner (Table 3).
Genomic DNA extraction and quantification: Genomic DNA was extracted from peripheral blood leukocytes using the phenol-chloroform extraction method following standard protocols. DNA purity and concentration were assessed using a NanoDrop spectrophotometer (Thermo Fisher Scientific, USA), and integrity was confirmed by electrophoresis on a 1% agarose gel.
Whole exome sequencing (WES): High-quality genomic DNA from affected individuals was subjected to whole exome sequencing (WES) using the Illumina NovaSeq 6000 platform (Illumina, San Diego, USA). Exome capture was performed with the Agilent SureSelect Human All Exon V7 kit, and sequencing reads were aligned to the human reference genome (GRCh38/hg38) using the Burrows-Wheeler Aligner (BWA-MEM). Variant calling was carried out using GATK v4.0, and annotation was performed with ANNOVAR and Ensembl Variant Effect Predictor (VEP). Identified variants were filtered based on quality parameters, population frequency (gnomAD, 1000 Genomes), and predicted pathogenicity.
Sanger sequencing validation: Putative pathogenic variants identified by WES were validated using Sanger sequencing. PCR primers were designed using Primer3 software, and amplified products were purified and sequenced bidirectionally using the ABI 3730xl DNA Analyzer (Applied Biosystems, USA). Sequencing chromatograms were analyzed with Chromas and aligned using BioEdit software to confirm segregation within families.
Bioinformatics analysis: The detected variants in SMPD1, HIP1R, and POMC genes were also confirmed by using online databases. These variants were not reported previously in any of the online databases, i.e., Human Gene Mutation Database, gnomAD, ClinVar, and Exome Variant Server.
RESULTS
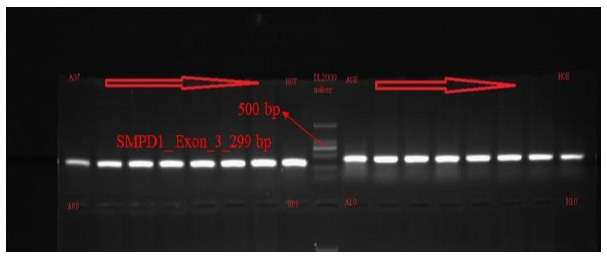
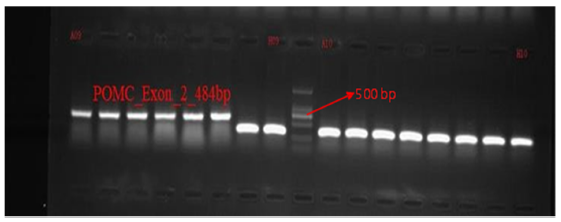
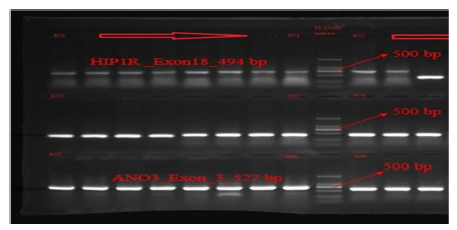
PCR amplification of the target exons of SMPD1, POMC, and HIP1R genes was successfully performed, producing fragments of the expected sizes: 299 bp for SMPD1 (exon 3), 484 bp for POMC (exon 2), and 494 bp for HIP1R (exon 18), shown in Fig. 4-6. The PCR products were purified and sequenced bidirectionally using the ABI 3730xl DNA Analyzer (Applied Biosystems, USA). For SMPD1, primers AGCACAGGAGGACCAGGA (forward) and GAGGTGCTCCAGCTCAACA (reverse) with Tm values of 59.3°C and 60.1°C, respectively, were used. A POMC amplification utilized TGGTCACAGTTTACCCACCA (forward) and TGAATCACGCCTATCACTGG (reverse) primers with Tm values of 59.8°C and 59.7°C, respectively. A HIP1R fragments were amplified using GGCTACATGGGAGACAGGAC (forward) and GAAGAAGCCGGACACAAAAG (reverse) primers with Tm values of 59.5°C and 59.9°C.
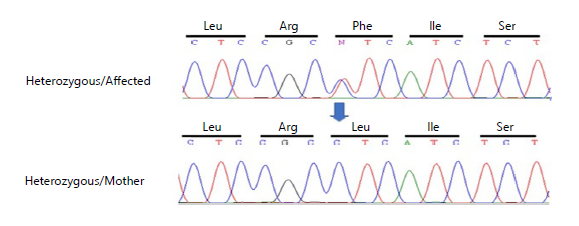
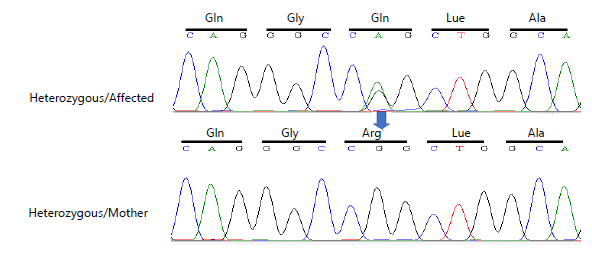
Whole exome sequencing (WES) was performed, using the DNA of an affected and normal member of the family to identify the variant. Subsequently, Sanger sequence analysis was performed to confirm the identified variant; a heterozygous missense substitution [c.1135C>T (p.Leu379Phe)] in exon 3 of the
|
|
|
|
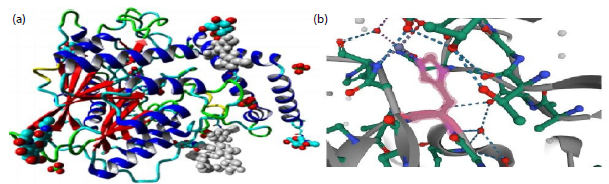
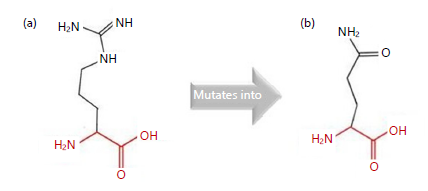
SMPD1 gene, manifested a change “T” into “A” in the DNA sequence. The chromatogram depicts that affected individuals were heterozygous while unaffected parents and siblings were homozygous (Fig. 7). The identified novel variant revealed that this nucleotide change led to the loss of function of the ASM protein product shown in Fig. 8(a-b). The current variant is located within a domain, the mutated residue is situated near a highly conserved region, and it affects the normal function of the ASM protein product. Structural effects were studied through online bioinformatics software HOPE and UniProt. Confirmation of the identified variant was performed by online bioinformatics tools, including MutationTaster, PolyPhen-2, and SIFT, which predicted that the current substitution produces a pathogenic altered protein. The variant was not reported previously in any of the online databases, i.e., Human Gene Mutation Database, gnomAD, ClinVar, and Exome Variant Server.
|
|
|
Whole exome sequencing (WES) was performed, using the DNA of an affected and a normal member of the family to identify the variant. Subsequently, Sanger sequence analysis was performed to confirm the identified variant [c.1802G>A (p. Arg601Gln)] in exon 18 of the HIP1R gene; the chromatogram depicts that unaffected parents and siblings were homozygous while affected individuals were heterozygous (Fig. 9). The identified variant [c.1802G>A (p.Arg601Gln)] revealed that this nucleotide change alters the function of the HIP1R protein product shown in Fig. 10(a-b), structural effects were studied through online bioinformatics software HOPE and UniProt. Confirmation of the identified variant was performed by online bioinformatics tools, including Mutation Taster, PolyPhen-2, and SIFT predicted that the current substitution produces a benign altered protein.
|
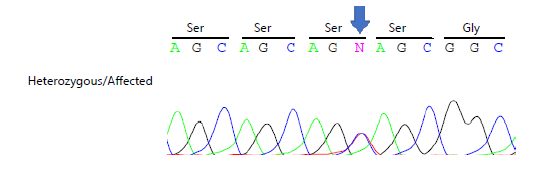
Whole exome sequencing (WES) was performed. Subsequently, Sanger sequence analysis was performed to confirm the identified heterozygous silent variant [c.282C>T (p. Ser94 Ser)] in exon 2 of the POMC gene, a change in nucleotide ‘C’ into ‘T’, but no amino acid change Fig. 11. The identified variant [c.282C>T (p. Ser94 Ser)] revealed no change in amino acid. This variant (rs28930368) was previously reported in online databases, i.e., Human Gene Mutation Database, gnomAD, ClinVar, and Exome Variant Server.
DISCUSSION
In this study, three major obesity-associated genes-SMPD1, HIP1R, and POMC were identified to segregate with obesity phenotypes in consanguineous families from Balochistan. These findings support the role of rare and inherited pathogenic variants in the development of monogenic obesity in populations with high consanguinity rates.
A heterozygous novel variant in SMPD1 was identified segregating with obesity and related clinical features, including type 2 diabetes, hypertension, and hyperphagia. A SMPD1 encodes acid sphingomyelinase, an enzyme responsible for sphingolipid metabolism. Dysregulation of sphingolipid pathways has previously been associated with metabolic disease and obesity, where high-fat diet exposure increases ceramide accumulation and leads to obesity, insulin resistance, and altered leptin signaling12. Pharmacological inhibition of SMPD1 activity has been shown to reverse diet-induced metabolic effects, highlighting its key role in obesity physiology13. Additionally, elevated secretory acid sphingomyelinase levels have been reported in obese children, reinforcing its clinical relevance to adiposity and metabolic dysfunction14. These findings collectively support the hypothesis that the SMPD1 variant identified in the present study may contribute to obesity susceptibility through disruption of lipid homeostasis.
The HIP1R missense variant detected in this study was also found to segregate in an autosomal dominant manner with obesity, hypertension, cardiovascular conditions, and diabetes. A HIP1R plays a role in clathrin-mediated endocytosis and interacts with metabolic pathways influencing lipid and glucose regulation15. Genome-wide association studies have linked HIP1R variants with increased BMI and altered metabolic signaling, suggesting its involvement in energy expenditure and adipocyte function16. Prior studies also report altered HIP1R gene expression patterns in diabetes, further supporting its pathogenic association with metabolic disorders17. The presence of this previously reported variant in the current cohort strengthens existing evidence linking HIP1R genetic variation with obesity pathogenesis.
Finally, a previously reported synonymous variant in POMC (p. Ser94Ser) was identified in a highly obese family. Although synonymous, prior studies have reported this variant among severely obese patients and implicated POMC dysfunction in neuroendocrine regulation of appetite and satiety18. A POMC plays a crucial role in hypothalamic melanocortin pathways controlling energy balance, and previously documented obesity-associated loci near POMC further support its importance in monogenic and polygenic obesity19. While no novel POMC variants have been reported previously in Pakistani cohorts, the presence of this variant in the current study confirms its relevance in this population20.
CONCLUSION
This study identified both novel and previously reported pathogenic variants in SMPD1, HIP1R, and POMC segregating with obesity in consanguineous families from Balochistan, expanding the mutational spectrum of obesity-related genes in underrepresented populations. The findings highlight the importance of population-specific genetic screening for early diagnosis, counseling, and management of hereditary obesity. Families without identified variants are candidates for whole-genome sequencing to uncover deep intronic or regulatory mutations. Future functional analyses of these candidate genes will provide deeper insight into the molecular mechanisms underlying obesity, informing targeted interventions and improving genotype-phenotype correlations in high-consanguinity communities.
SIGNIFICANCE STATEMENT
This study reports the identification of novel and known pathogenic variants in the SMPD1, HIP1R, and POMC genes among consanguineous families from Balochistan with early-onset obesity. The findings contribute to understanding the genetic architecture of obesity in underrepresented populations and emphasize the need for population-specific genetic screening programs. This work provides valuable insight for future diagnostic strategies, genetic counselling, and targeted management of obesity in Pakistan’s high-consanguinity communities.
ACKNOWLEDGMENT
We thank the families for their cooperation and participation in this study.
REFERENCES
- Pi-Sunyer, X., 2009. The medical risks of obesity. Postgrad. Med., 121: 21-33.
- WHO, 2025. Obesity and Overweight. World Health Organization, Geneva, Switzerland.
- Herrera, B.M. and C.M. Lindgren, 2010. The genetics of obesity. Curr. Diabetes Rep., 10: 498-505.
- Hruby, A. and F.B. Hu, 2015. The epidemiology of obesity: A big picture. PharmacoEconomics, 33: 673-689.
- Locke, A.E., B. Kahali, S.I. Berndt, A.E. Justice and T.H. Pers et al., 2015. Genetic studies of body mass index yield new insights for obesity biology. Nature, 518: 197-206.
- Silventoinen, K., A. Jelenkovic, R. Sund, Y.M. Hur and Y. Yokoyama et al., 2016. Genetic and environmental effects on body mass index from infancy to the onset of adulthood: An individual-based pooled analysis of 45 twin cohorts participating in the collaborative project of development of anthropometrical measures in twins (CODATwins) study. Am. J. Clin. Nutr., 104: 371-379.
- Krude, H., H. Biebermann, W. Luck, R. Horn, G. Brabant and A. Grüters, 1998. Severe early-onset obesity, adrenal insufficiency and red hair pigmentation caused by POMC mutations in humans. Nat. Genet., 19: 155-157.
- Kühnen, P., K. Clément, S. Wiegand, O. Blankenstein and K. Gottesdiener et al., 2016. Proopiomelanocortin deficiency treated with a melanocortin-4 receptor agonist. N. Engl. J. Med., 375: 240-246.
- Simonaro, C.M., R.J. Desnick, M.M. McGovern, M.P. Wasserstein and E.H. Schuchman, 2002. The demographics and distribution of type B Niemann-Pick disease: Novel mutations lead to new genotype/phenotype correlations. Am. J. Hum. Genet., 71: 1413-1419.
- Metzler, M., B. Li, L. Gan, J. Georgiou, C.A. Gutekunst and Y. Wang et al., 2003. Disruption of the endocytic protein HIP1 results in neurological deficits and decreased AMPA receptor trafficking. EMBO J., 22: 3254-3266.
- Saeed, S., T.A. Butt, M. Anwer, M. Arslan and P. Froguel, 2012. High prevalence of leptin and melanocortin-4 receptor gene mutations in children with severe obesity from Pakistani consanguineous families. Mol. Genet. Metab., 106: 121-126.
- Boini, K.M., C. Zhang, M. Xia, J.L. Poklis and P.L. Li, 2010. Role of sphingolipid mediator ceramide in obesity and renal injury in mice fed a high-fat diet. J. Pharmacol. Exp. Ther., 334: 839-846.
- Lopez, X., A.B. Goldfine, W.L. Holland, R. Gordillo and P.E. Scherer, 2013. Plasma ceramides are elevated in female children and adolescents with type 2 diabetes. J. Pediatr. Endocrinol. Metab., 26: 995-998.
- Farooqi, I.S., G. Matarese, G.M. Lord, J.M. Keogh and E. Lawrence et al., 2002. Beneficial effects of leptin on obesity, T cell hyporesponsiveness and neuroendocrine/metabolic dysfunction of human congenital leptin deficiency. J. Clin. Invest., 110: 1093-1103.
- Bradley, S.V., T.S. Hyun, K.I. Oravecz-Wilson, L. Li and E.I. Waldorff et al., 2007. Degenerative phenotypes caused by the combined deficiency of murine HIP1 and HIP1r are rescued by human HIP1. Hum. Mol. Genet., 16: 1279-1292.
- Turcot, V., Y. Lu, H.M. Highland, C. Schurmann and A.E. Justice et al., 2018. Protein-altering variants associated with body mass index implicate pathways that control energy intake and expenditure in obesity. Nat. Genet., 50: 26-41.
- Takematsu, E., A. Spencer, J. Auster, P.C. Chen, A. Graham, P. Martin and A.B. Baker, 2020. Genome wide analysis of gene expression changes in skin from patients with type 2 diabetes. PLoS ONE, 15.
- Challis, B.G., L.E. Pritchard, J.W.M. Creemers, J. Delplanque and J.M. Keogh et al., 2002. A missense mutation disrupting a dibasic prohormone processing site in pro-opiomelanocortin (POMC) increases susceptibility to early-onset obesity through a novel molecular mechanism. Hum. Mol. Genet., 11: 1997-2004.
- Speliotes, E.K., C.J. Willer, S.I. Berndt, K.L. Monda and G. Thorleifsson et al., 2010. Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nat. Genet., 42: 937-948.
- Shabana and S. Hasnain, 2016. The p. N103K mutation of leptin (LEP) gene and severe early onset obesity in Pakistan. Biol. Res., 49.
How to Cite this paper?
APA-7 Style
Daud,
S., Naudhani,
S., Malik,
S.F., Panezai,
H., Iqbal,
F. (2026). Whole-Exome Sequencing Identifies Pathogenic Variants in SMPD1, HIP1R, and POMC Associated with Obesity in a Consanguineous Pakistani Population. Asian Journal of Emerging Research, 8(2), 96-105. https://doi.org/10.21124/ajer.2026.96.105
ACS Style
Daud,
S.; Naudhani,
S.; Malik,
S.F.; Panezai,
H.; Iqbal,
F. Whole-Exome Sequencing Identifies Pathogenic Variants in SMPD1, HIP1R, and POMC Associated with Obesity in a Consanguineous Pakistani Population. Asian J. Emerg. Res 2026, 8, 96-105. https://doi.org/10.21124/ajer.2026.96.105
AMA Style
Daud
S, Naudhani
S, Malik
SF, Panezai
H, Iqbal
F. Whole-Exome Sequencing Identifies Pathogenic Variants in SMPD1, HIP1R, and POMC Associated with Obesity in a Consanguineous Pakistani Population. Asian Journal of Emerging Research. 2026; 8(2): 96-105. https://doi.org/10.21124/ajer.2026.96.105
Chicago/Turabian Style
Daud, Shakeela, Sara Naudhani, Sobia Faisal Malik, Hameeda Panezai, and Fatima Iqbal.
2026. "Whole-Exome Sequencing Identifies Pathogenic Variants in SMPD1, HIP1R, and POMC Associated with Obesity in a Consanguineous Pakistani Population" Asian Journal of Emerging Research 8, no. 2: 96-105. https://doi.org/10.21124/ajer.2026.96.105

This work is licensed under a Creative Commons Attribution 4.0 International License.