
Protonation in Benzyl Alcohol: Different Proton Affinities, Same Neutral Molecule
-
Oko Emmanuel Godwin
Department of Chemical Sciences, Federal University Wukari, PMB 1020, Wukari, Taraba State, Nigeria
Emmanuel E. Etim
Department of Chemical Sciences, Federal University Wukari, PMB 1020, Wukari, Taraba State, Nigeria
Sulaiman Adeoye OlagboyeDepartment of Chemistry, Ekiti State University, Ado -Ekiti, Nigeria
M.E. KhanDepartment of Chemistry, Federal University Lokoja, Kogi State, Nigeria
| Received 16 Oct, 2020 |
Accepted 01 Jan, 2021 |
Published 31 Mar, 2021 |
ABSTRACTBackground and Objectives: Benzyl Alcohol (BA) has 6 possible protonated sites which give rise to 6 possible proton affinities for the same neutral molecule (BA). However, experimentally, only one proton affinity value is reported for BA but computationally, each possible protonated site can be specifically optimized and the proton affinity calculated for each protonated species. Materials and Methods: In this study, quantum chemical calculations were carried out using GAUSSIAN 09 suite of programs. Five (5) different computational methods have been employed to calculate the proton affinity for each possible protonated analogue. Results: The results obtained are compared with the experimental value in order to determine the protonated analogue that corresponds to the experimental value and which is thermodynamically more favored. It was found that the best site for the protonation of BA is via the C 7 atom as it corresponds more to the experimental value and also found to be more stable. Conclusion: Generally, the best computational methods for the calculation of the PA values of BA was observed to be B3LYP/6-311++G** but on the basis of the best site of protonation G 4 method happened to be the best computational method.
INTRODUCTION
Benzyl Alcohol (also known as benzenemethanol, alpha-toluenol or phenylcarbinol) is a colorless aromatic alcohol consisting of benzene with a hydroxymethyl substituent with a molecular formula of C6H5CH2OH or C7H8O as shown in Fig.1 below.
BA finds application as solvents for paints, waxes, inks, Lacquers, shellacs, dyes and resins. As Fragrance and preservatives in soap, perfume, e-cigarettes.it also acts as insects repellants and a developer for photographic films.BA also finds application in health1,2. It several usefulness have been attributed to its polarity, low toxicity, low vapor pressure and solubility in other solvents like water, alcohols and diethylether.
BA can be prepared or produced via the following path (a) Hydrolysis of toluene via benzyl chloride, shown in Eq. 13. (b) Hydrogenation reaction of benzaldehyde Eq. 2 and (c) The reaction of phenylmagnesium bromide with formaldehyde (Grignard reagent)4
$$
\mathrm{C}_{5} \mathrm{H}_{5} \mathrm{Cl}+\mathrm{H}_{2} \mathrm{O} \longrightarrow \mathrm{C}_{6} \mathrm{H}_{5} \mathrm{CH}_{2} \mathrm{OH}+\mathrm{HCl}
$$ |
(1) |
$$
\mathrm{C}_{6} \mathrm{H}_{5} \mathrm{CHO} \quad \mathrm{H}_{2} \longrightarrow \mathrm{C}_{6} \mathrm{H}_{5} \mathrm{CH}_{2} \mathrm{OH}
$$ |
(2) |
Just like other alcohols, BA is a precursor for most esters as it undergoes esterification reaction with carboxylic acid to form esters, Eq. 35.
$$
\mathrm{PhCH}_{2} \mathrm{OH}+\text { HOAccatalyst }{\longrightarrow} \mathrm{PhCH}_{2} \mathrm{OAc}
$$ |
(3) |
It undergoes deprotonation reaction to form a benzylate anion and also undergoes a Ritter reaction with acrylonitrile to yield benzyl acrylamide as shown in Eq. 46.
$$
\mathrm{C}_{6} \mathrm{H}_{5} \mathrm{CH}_{2} \mathrm{OH}+\mathrm{CH}_{2}=\mathrm{CHCN} \longrightarrow \mathrm{C}_{6} \mathrm{H}_{5} \mathrm{CH}_{2} \mathrm{NHCOCH}=\mathrm{CH}_{2}
$$ |
(4) |
Protonation simply refers to the resultant change in charge and mass of an atom, molecule or an ion brought about by the addition of a proton. The phenomenon "protonation" is quite different from "hydrogenation" in which the charge of the protonated specie is unaffected and so should not be misunderstood for protonation.
Proton Affinity (PA, Epa) is the energy (Enthalpy) released in a reaction between a specie (anion, neutral atom or molecule) and a proton in the gas phase. This entails that when proton is added to specie in gas phase (protonation), energy is released (proton affinity). It is usually represented by the general Eq. 5:
$$
\mathrm{PA}(\mathrm{M})=\Delta_{\mathrm{f}} \mathrm{H}^{0}(\mathrm{M})+\Delta_{\mathrm{f}} \mathrm{H}^{0}\left(\mathrm{H}^{+}\right)-\Delta_{\mathrm{f}} \mathrm{H}^{0}\left(\mathrm{MH}^{+}\right)
$$ |
(5) |
Where ΔfH0 represent enthalpies of formation7.
The relationship between the PA of an acid /base and its strength was reportedly recorded by Poad8 were they pointed out that if in the gas phase the proton affinity is higher, the base will be stronger while the conjugate acid will be weaker. The finding included that the strongest known base is Orthodiethynylbenzenedianion with an Epa = 1843 kJ mol-1 and Hunter and Lias9 recorded the weakest known base, helium atom with Epa= 177.8 kJ mol-1. It's quite obvious that homo-nuclear species like O2,H2 have just one PA, hetero-nuclear species can have more than one PA e.g. SiO3 with two possible PAs, SCO with three protonation sites, as reported by a team led by Etim et al.10,11, Toluene with four PAs12 and phenol with two PAs13,14.
One great application of PA in modern sciences lies in our quest to understand and explore the interstellar medium (ISM), one way of achieving this is by the study of the PAs of such molecules since their presence have been detected in the ISM15,16,10,11.
Two basic methods are known for the determination of the PA of specie; experimental and computational approaches:
Experimental Approaches: This is an objective scientific known method which uses kinetic or thermodynamic data such asenthalpy change and the equilibrium constants in a proton transfer reaction. Some of the measurement techniques employed in this method include; High Pressure Mass Spectroscopy (HPMS)17, Ion Mobility Spectrometry (IMS)18,19, Flame Ion Mass Spectrometry (FIMS), Selected Ion Flow Tubes (SIFT), Iion Cyclotron Resonance (ICR) Spectrometry and Knudsen Cell Mass Spectrometry (KCMS)20. HPMS, SIFT, ICR are suitable for determining the PA of low temperature molecular species e.g. organic compounds. KCMS is suitable for metallic compounds which decomposes at a lesser temperature while FIMS for metallic compounds which decomposes at a high temperature.
Computational Approach: Owing to difficulties in obtaining PAs experimentally gives rise to the computational approach. Uses the Ab-initio methods (which is based on quantum mechanics) or semi-empirical method to used to obtain the PA via computer simulations. The Ab-initio method is an approximate chemical calculation based on theoretical principles independent of experimental data which enables solution of chemical parameters of the Schrodinger equation e.g. electron density or energy from which PA is calculated. Different types of Ab-initio methods such as HF/6-311++G**, B3LYP/6-311++G**, MP2/6-311++G**, MP2/cc-pVDZ, G421 have enabled researchers predicted structures, interactions and properties of systems accurately or approximately. For examples, Etim10,11 computationally predicted the PA of SO3 to be 140.2 kcal mol-1 using the B3LYP/6-311++G** level of theory, meanwhile the experimental value is 140.6 kcal mol-1. In their Ab-initio study of carbonylsulfide CSO. They again made a very perfect prediction using the HF/6-311++G** method when compared to the experimental values where they predicted the PA of CSO to be 150.7 kcal mol-1 and the experimental value is 150.2 kcal mol-1.
These findings support the authenticity and accuracy of the Ab-initio methods and calls for their use in comparison with experimental data as a tool in deciding which could serve as an alternative, thus the aim of this work; BA, a molecule with six (6) possible protonated species, which one correspond to the reported experimental value? We therefore aim to specify the best site of protonation as the experimental procedure is deficient of such information.
MATERIALS AND METHODS
All the electronic structure calculations (thermodynamic properties) for the required proton affinities were implemented using the Gaussian 09 suit of programs22. Five (5) Ab-initio computational methods which include: Gaussian 04 (G4) compound method, Hartree-Fock (HF) method, Becke, three-parameter, Lee-Yang-Parr (B3LYP) method, Moller-Plesset perturbation theory (MP2) at 6-311++G** bias set and Moller-Plesset perturbation theory (MP2) at cc-pVDZ basis set were used in carrying out the calculations.
These methods were chosen based on experience from our previous studies15,16,10,11 in which these methods were applied and accurate results were obtained. Also, the use of these methods is essential in monitoring how consistent or coherent the results are which will aid in determining the best method. The proton affinity (PA) is calculated as the difference in energy (Electronic Energy) between a neutral specie and its protonated analogue.
|
|||||||||||||||
| Molecule |
Protonation site
|
||||||||||||||
| BA (C7H8O) | Proton attached to O atom (kcal mol-1) | Proton attached to C7 atom (kcal mol-1) | Proton attached to C2 atom (kcal mol-1) | Proton attached to C4 atom (kcal mol-1) | Proton attached to C6 atom (kcal mol-1) | Proton attached to C5 atom (kcal mol-1) | |||||||||
| X | PA | Error | PA | Error | PA | Error | PA | Error | PA | Error | PA | Error | |||
| 1 | 195.5 | 9.5 | 180.5 | -5.4 | 188.2 | 2.2 | 195.1 | 9.1 | 190.8 | 4.8 | 196.1 | 10.1 | |||
| 2 | 199.3 | 13.3 | 182.5 | 3.5 | 183.0 | 3.0 | 191.4 | 5.4 | 187.2 | 1.2 | 191.4 | 5.4 | |||
| 3 | 191.5 | 5.5 | 174.5 | -11.5 | 175.0 | -11.0 | 180.9 | -5.1 | 177.8 | -8.2 | 180.9 | -5.1 | |||
| 4 | 194.9 | 8.9 | 178.2 | -7.8 | 178.0 | -8.0 | 184.2 | -1.8 | 180.7 | -5.2 | 184.2 | -1.8 | |||
| 5 | 200.4 | 14.4 | 189.0 | 3.0 | 188.7 | 2.7 | 197.3 | 11.3 | 192.9 | 6.9 | 197.4 | 11.4 | |||
| 6 | 186.0 | NA | 186.0 | NA | 186.0 | NA | 186.0 | NA | 186.0 | NA | 186.0 | NA | |||
| XMethod, 1HF/6-311++G**, 2B3LYP/6-311++G**, 3MP2/6-311++G**, 4MP2/cc-pVDZ, 5G4, 6Expt | |||||||||||||||
|
|||||||||||||||
| Parameter | Energy (Hartree/Particle)* | ||||||||||||||
| Protonation at O1 atom | Protonation at C7 atom | Protonation at C2 atom | Protonation at C4 atom | Protonation at C6 atom | Protonation at C5 atom | ||||||||||
| Zero-point correction | 0.154046 | 0.152069 | 0.153613 | 0.152478 | 0.152694 | 0.152572 | |||||||||
| Thermal correction to Energy | 0.161362 | 0..160296 | 0.160694 | 0.159785 | 0.160004 | 0.159848 | |||||||||
| Thermal correction to enthalpy | 0.162307 | 0.161240 | 0.161638 | 0.160729 | 0.160948 | 0.160793 | |||||||||
| Thermal correction to Gibss Free Energies | 0.122304 | 0.117289 | 0.122042 | 0.120444 | 0.120916 | 0.120869 | |||||||||
| Sum of electronic & zero-point Energies | -344.848 | -344.824 | -344.836 | -344.847 | -344.840 | -344.848 | |||||||||
| Sum of electronic & thermal Energies | -344.840 | -344.816 | -344.829 | -344.839 | -344.833 | -344.841 | |||||||||
| Sum of electronic & thermal Enthalpy | -344.839 | -344.815 | -344.828 | -344.839 | -344.832 | -344.840 | |||||||||
| Sum of electronic & thermal Free Energies | -344.879 | -344.859 | -344.868 | -344.879 | -344.872 | -344.880 | |||||||||
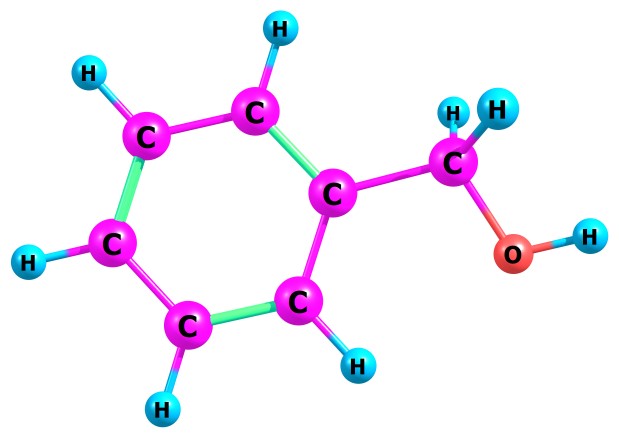
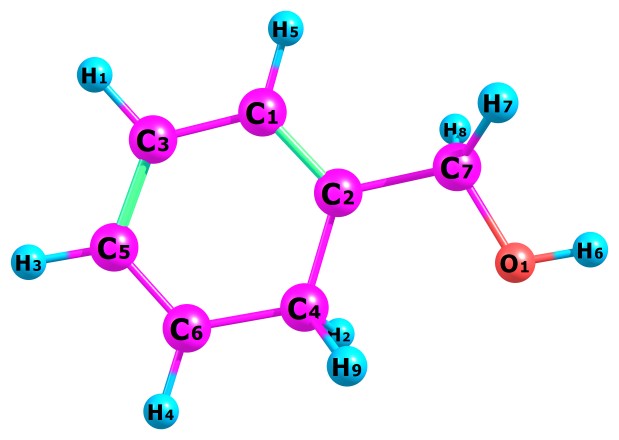
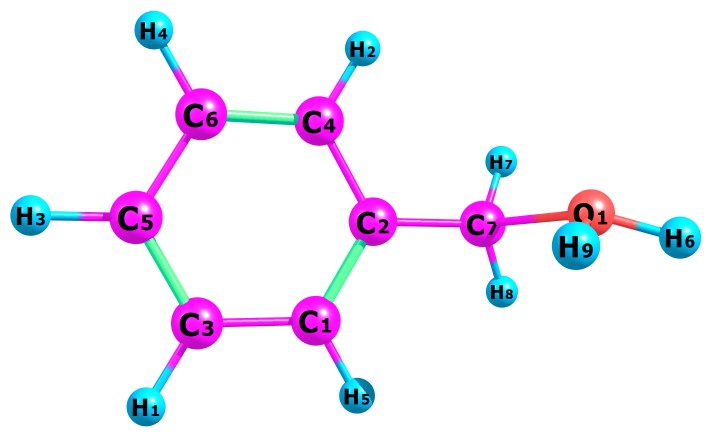
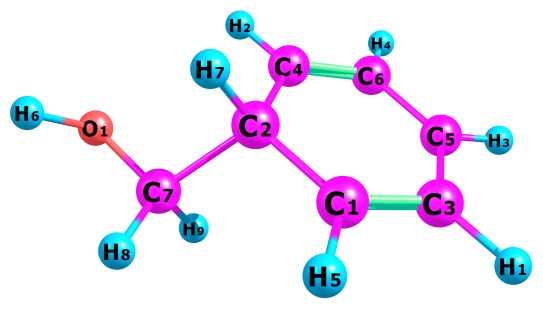
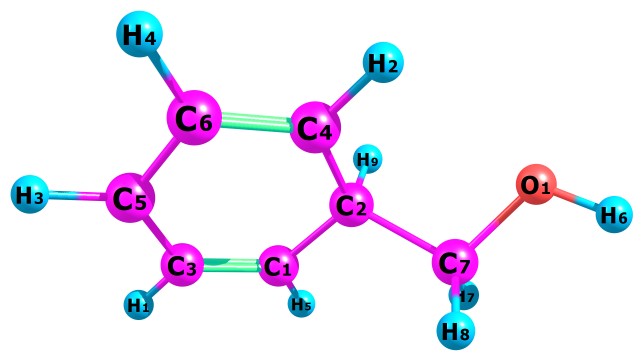
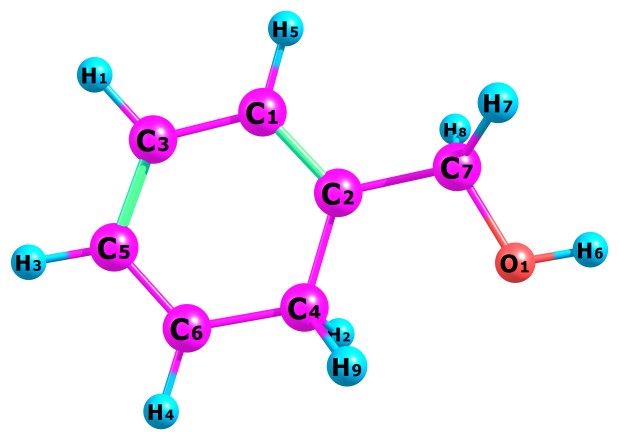
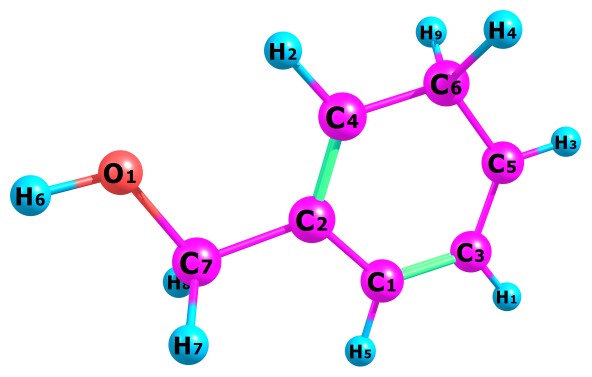
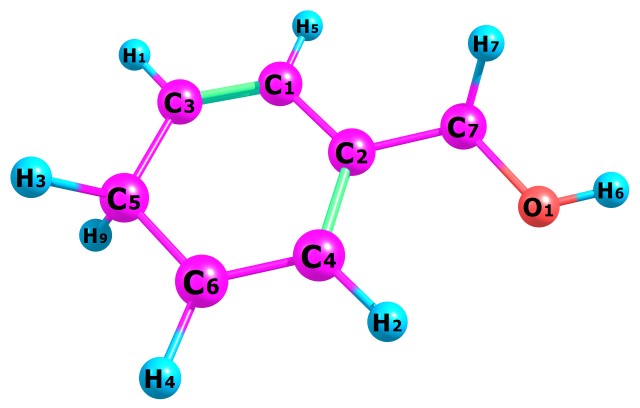
Table 1 present our calculated Proton Affinities (PAs) of Benzyl Alcohol (BA) in collaboration with its observed experimental value (186.0 kcal mol-1) reported by the NIST website to aid our comparison. Fig.2a-b and Fig.3a-f depicts the optimized geometry of BA and its protonated analogues. BA has six possible protonated sites, giving rise to six possible proton affinities and six possible protonated analogues (species). The possible sites for the attachments of protons are: Proton attachment to O atom, to C2 atom, to C4 atom, to C5 atom, to C6 atom and to C7 atom. C1 an C3 are not independently recorded as individual protonated site due to equivalency, C6 is equivalent to C3 and C4 is equivalent to C1.
- Proton attachment to O atom: The second order Moller-plesset perturbation theory MP2 at 6-311G** levof theory recorded the least error out of the 5 computational methods applied when proton was attached to O atom. All values obtained from the 5 computational methods were a little bit higher than the experimental values of 186.0 kcal mol-1, only the MP2/6-311++G** method gave a value (191.5 kcal mol-1) more chosen to that of the experimental, an error of 5.5 kcal mol-1 was recorded which happens to be the least of all the methods making it (MP2/6-311++G**) the method of choice for calculating PA in BA when proton is attached to the O atom. Generally, all the errors recorded where large. Only one method (MP2/6-311++G**) seem better.
- Proton attached to C7 atom: With respect to the five computational methods under investigation, G4 method obtained the best prediction with a PA value (189.0 kcal mol-1) with just 3units higher than the experimental value. Proton attachment to C7 has a good level of precision from all the computational methods. Generally, errors here too were large. Three of the five methods obtained better predictions (HF/6-311++G**, G4 and B3LYP/6-311++G**) gave the least error. This protonated analogue appeared more stable than via O atom.
- Proton attached to C2 atom: On this site of protonation, there was also a high precision or closeness of all the computational methods to the experimental value. Three out of five of the methods including B3LYP/6-311++G**, G4 and HF/6-311++G** gave the least errors in that order and as such giving the HF method priority when it comes to attaching proton at the C2 atom in BA. This specie appeared to be stable too.
- Proton attached to C4: Two methods (MP2/cc-pVDZ and MP2/6-311++G**) happen to be the only methods with a prediction a bit closer to the experimental value (i.e 184.2 kcal mol-1 and 180.9 kcal mol-1 respectively). This specie appeared to be less stable.
- Proton attached to C6 atom: Generally, from all the computational methods applied in this work and from all calculations made on the various possible protonated sites, the B3LYP/6-311++G** gave the best prediction (187.2 kcal mol-1) at this site compared to experimental value of 186.0 kcal mol-1. A least error of 1.2 kcal mol-1 was recorded while the other computational methods applied on this site of protonation gave predictions that are a bit away from the experimental value, thereby altering stability on this site.
- Proton attached to C5 atom: Just as shown for C4, PAs obtained when proton is attached to C6 gave values that are far more away from the experimental value when compared to when proton is attached to other sites. The value predicted by MP2/CC (184.2) happens to be the only value closer to the experimental (186.0) with an error of 1.8 in magnitude. A critical study of stability via the value shows protonation on this site to be very much less stable.
From the above proceedings (discussion on the six possible protonated site with respect to the result obtained from the 5 different methods and their errors), we have elucidated and simplified the process of understanding our findings that the B3LYP/6-311++G** gave a better prediction of the PA of BA at the C6 analogue compared to every other computational methods implemented (with a prediction of 187.2 kcal mol-1 as against 186 kcal mol-1 of the experimental value). On the basis of the protonated analogues or site of protonation, PAs obtained when proton is attached to O, C4, C5 and C6 were unduly wide away from the experimental value, this entails the difficulty of protonation and the unstable nature of those protonated analogues, thus leaving us with species with proton at C7 and C2. But a critical look at Table 2, we noticed that protonation at C7 was more favored than protonation at C2 atom, this reveal that PA was best predicted when proton is attached to the C7 atom since it is thermodynamically more stable and has values closer to the experimental observed value implying that both experimental and PA values for the specie correspond more. The results from our calculated PA values for BA therefore reveals that protonation via carbon atom (A site that is less electronegative and with less electron density) is better than protonation via the oxygen atom (which is more electronegative and with more electron density. As can be seen in Table 2a stability checks shows that out of the five carbon sites, protonation at the C7 atom was more favored and PA corresponds to a site that is thermodynamically more favored23. In extension, this finding also supports our previous work on Carbonyl sulfide CSO that electro negativity is not a good standard of judgment in predicting the protonation site of a molecule10.
CONCLUSION
Five different computational methods have been applied in studying the protonation of Benzyl Alcohol (BA) with the aim to figure out the best site of protonation. the results obtained from the five different computational methods and their errors coupled with their thermodynamic stability checks reveals that the best site of protonation is through Carbon atom particularly the C7 atom which is less electronegative and with less electron density since this protonated analogue was found to be more stable and corresponds to the experimentally obtained value, and that electronegativity is not a good standing point in predicting the site of protonation of a molecule.
REFERENCES
- European Committee, 2002. Opinion of the scientific committee on food on benzyl alcohol. Scientific committee on food. SCF/CS/ADD/FLAV/78 Final. 17 Sept, 2002.
- Ash, H., 2004. Handbook of preservatives. Synapse Info Resources, United States
- Brühne, F. and E. Wright, 2000. Benzyl alcohol. Ullmann's Encyclopedia of Industrial Chemistry, Weinheim: Wiley-VCH.
- Hao, Y., C. Pischetola, F. Cárdenas-Lizana and M.A. Keane, 2020. Selective liquid phase hydrogenation of benzaldehyde to benzyl alcohol over alumina supported gold. Catal. Lett., 150: 881-887.
- Baek, H., M. Minakawa, Y.M.A. Yamada, J.W. Han and Y. Uozumi, 2016. In-water and neat batch and continuous-flow direct esterification and transesterification by a porous polymeric acid catalyst. Sci. Rep., Vol. 6: 25925.
- Yamato, T., J.Y. Hu and N. Shinoda, 2007. Perfluorinated sulfonic acid resin (Nafion-H) catalysed ritter reaction of benzyl alcohols. J. Chem. Res., 2007: 641-643.
- Holmes, J.L., N.A. van Huizen and P.C. Burgers, 2017. Proton affinities and ion enthalpies. Eur. J. Mass Spectrom., 23: 341-350.
- Poad, B.L., N.D. Reed, C.S. Hansen, A.J. Trevitt and S.J. Blanksby et al., 2016. Preparation of an ion with the highest calculated proton affinity: Ortho-diethynylbenzene dianion. Chem. Sci., 7: 6245-6250.
- Hunter, E.P.L. and S.G. Lias, 1998. Evaluated gas phase basicities and proton affinities of molecules: An update. J. Phys. Chem. Ref. Data, 27: 413-656.
- Etim, E.E., G.E. Oko, A.I. Onen and O.A. Ushie et al., 2018. Computaational studies of sulphur trioxide (SO3) and its protonated analogues. J. Chem. Soc. Nigeria, 43: 10-17.
- Etim, E.E., C. Andrew, U. Lawal, I.S. Udegbunam and E.G. Ukpong, 2019. Protonation of carbonyl sulfide: ab initio study. J. Appl. Sci., 20: 26-34.
- Devlin III, J.L., J.F. Wolf, R.W. Taft and W.J. Hehre, 1976. The proton affinities of toluene. J. Am. Chem. Soc., 98: 1990-1992.
- Eckert-Maksic, M., M. Klessinger and Z.B. Maksic, 1995. Theoretical calculations of proton affinities in phenol. Chem. Phys. Lett., 232: 472-478.
- DeFrees, D.J., R.T. McIver and W.J. Hehre, 1977. The proton affinities of phenol. J. Am. Chem. Soc., 99: 3853-3854.
- Etim, E.E. and E. Arunan, 2016. Interstellar isomeric species: Energy, stability and abundance relationship. Eur. Phys. J. Plus, 131: 448
- Etim, E.E., B.S. Abah, I.E. Mbakara, E.J. Inyang and O.P. Ukafia, 2017. Quantum chemical calculations on silicon monoxide (SiO) and its protonated analogues. Trop. J. Applied Nat. Sci., 2: 61-68.
- Kebarle, P., 1977. Ion thermochemistry and salvation from gas phase ion equilibria. Annual Rev. Phys. Chem., 28: 445-476.
- Tabrizchi, M. and S. Shooshtari, 2003. Proton affinity measurements using ion mobility spectrometry. J. Chem. Thermodyn., 35: 863-870.
- Eiceman, G.A., Z. Karpas and H.H. Hill, Jr. 2014. Ion mobility spectrometry. 3rd Edn., CRC Press, United States.
- Chen, Q. and J.M. Goodings, 1998. The measurement of high proton affinities for a variety of metallic compounds: a new approach by flame-ion mass spectrometry. Int. J. Mass Spectrom., 181: 181-199.
- Cramer, C.J., 2002. Essentials of Computational Chemistry Theories and Models. John Wiley & Sons Ltd., New York.
- Frisch, M.J., M.J. Frisch, G.W. Trucks, H.B. Schlegel and G.E. Scuseria et al., 2010. Gaussian 09, Revision D. 01. Gaussian Inc., Wallingford, CT.
- Tu, Y.P., 2006. Dissociative protonation sites: Reactive centres in protonated molecules leading to fragmentation in mass spectrometry. J. Org. Chem., 71: 5482-5488.
How to Cite this paper?
APA-7 Style
Godwin,
O.E., Etim,
E.E., Olagboye,
S.A., Khan,
M.E. (2021). Protonation in Benzyl Alcohol: Different Proton Affinities, Same Neutral Molecule. Asian Journal of Emerging Research, 3(1), 19-24. https://doi.org/10.3923/AJERPK.2021.19.24
ACS Style
Godwin,
O.E.; Etim,
E.E.; Olagboye,
S.A.; Khan,
M.E. Protonation in Benzyl Alcohol: Different Proton Affinities, Same Neutral Molecule. Asian J. Emerg. Res 2021, 3, 19-24. https://doi.org/10.3923/AJERPK.2021.19.24
AMA Style
Godwin
OE, Etim
EE, Olagboye
SA, Khan
ME. Protonation in Benzyl Alcohol: Different Proton Affinities, Same Neutral Molecule. Asian Journal of Emerging Research. 2021; 3(1): 19-24. https://doi.org/10.3923/AJERPK.2021.19.24
Chicago/Turabian Style
Godwin, Oko, Emmanuel, Emmanuel E. Etim, Sulaiman Adeoye Olagboye, and M. E. Khan.
2021. "Protonation in Benzyl Alcohol: Different Proton Affinities, Same Neutral Molecule" Asian Journal of Emerging Research 3, no. 1: 19-24. https://doi.org/10.3923/AJERPK.2021.19.24

This work is licensed under a Creative Commons Attribution 4.0 International License.