
Microtubule Targeted Pharmacophore Study of Cryptophycin Derivatives for Effective Treatment of Cancer
-
Appavoo Umamaheswari
Department of Pharmaceutical Technology, University College of Engineering (BIT Campus), Anna University, Tiruchirappalli, India
Sakthivel Lakshmana Prabu
Department of Pharmaceutical Technology, University College of Engineering (BIT Campus), Anna University, Tiruchirappalli, India
Ayarivan PuratchikodyDepartment of Pharmaceutical Technology, University College of Engineering (BIT Campus), Anna University, Tiruchirappalli, India
| Received 25 Feb, 2020 |
Accepted 31 Dec, 2020 |
Published 03 Jun, 2021 |
Background and Objectives: Worldwide in 21st century cancer become the foremost cause of death and it is become the second leading cause of death also estimated that 1 in 6 deaths is due to cancer. Recently, cryptophycin is a microtubule agent has been used in the treatment of cancer. This study was aimed to develop a pharmacophore model for cryptophycin analogues of tubulin inhibitors by using quantitative approach to retrieve structurally diverse compounds with desired biological activity. Materials and Methods: Silicon graphics octane 2 R12000 computer running lrix 6.5.12 with Catalyst version 4.6 was used to generate pharmacophore models. A training set of 20 compounds and test set of 18 compounds were selected from the database was formed using HypoGen model based on the quantitative predictive character such as structural diversity and biological activity against human leukemia CCRF-CEM tumor cell lines. Results: A training set of 20 cryptophycin inhibitors exhibiting in vitro IC50 values ranging from 0.04 nm to 2500 nm were utilized to generate a quantitative pharmacophore. The best pharmacophore hypothesis generation consists of one hydrogen bond acceptor, one hydrogen bond donor, one hydrophobic aliphatic and one ring aromatic as important features. The selected best pharmacophore hypothesis yielded a RMS deviation of 0.774 and a correlation coefficient of 0.948 with a cost difference of 57.71. Predicted accuracy value for the test set compounds were greater than 95%. Conclusion: The results of this study showed distinct chemical features useful in the design and development of inhibitors with greater selectivity towards tubulin.
INTRODUCTION
The major medical issue in this 21st century is the extreme increase and incidence of cancer and it has become the foremost cause of death. As per WHO report in 2018, the death due to cancer is 9.6 million and it became the second leading cause of deaths1. Worldwide, the most common cause of cancer death occurs in lung (1.76 million deaths), liver (782000 deaths), colorectal (862000 deaths), stomach (783000 deaths) and breast (627000 deaths). There is a steep increase in cancer death by 2019, and more than 1.7 million new cancer cases are probable to be diagnosed2. In the next two decades, the number of case is expected to rise by 70% stipulating that cancer will surpass heart diseases3. It is an alarm to the research scientist to identify and develop new anticancer agents to fulfill the current therapeutic needs.
A major goal in cancer chemotherapy is that the drugs should target efficiently against tumor cells rather than normal cells. Eukaryotic cellular process that includes cell growth and division, motility, intracellular transport, functioning as part of the spindle to ensure proper chromosome segregation, cell division and adapting different shapes to interact with environment are all governed by polymerization dynamics in microtubules4. The polymerization of microtubules occurs by a reversible nucleation-elongation mechanism, non-covalent addition of α and β tubulin heterodimers at both ends of microtubules5. The two ends of a microtubule are not equivalent. These microtubules are hollow, cylindrical structures built up of multifunctional cytoskeletal proteins as a helical array which consists of two similar subunits such as α and β tubulin heterodimers as an alternative. The cylinder consists of thirteen rows of tubulin heterodimers. Among the various tubulin heterodimers, eight β-tubulin isotypes are identified in human. Over appearance of class III β-tubulin among these eight β-tubulin isotypes is an indicator of confrontation to tubulin targeting agents6. During mitosis phase, microtubules facilitate the replication of chromosomes into two identical groups. Phases involved in the formation of identical group of chromosome are prometaphase, metaphase, anaphase and telophase. Each single chromosome should attach to a bipolar spindle of microtubule, if there occurs any disturbance or failure or stop of the attachment of chromosome with microtubules, it leads to cell death7,8.
Microtubule targeting agent is a category of drug used in the treatment of cancer9. These categories of drugs bind either with the tubulin or microtubulin which depends on specific location of the binding sites10. These microtubule targeting agents bind to five known binding sites like laulimalide, taxane/epothilone, vinca alkaloid, maytansine and colchicine binding site and disturb the microtubulin and induces cell cycle arrest in the G2/M phase which ultimately fetches out microtubules, an attractive target for drug discovery3,11. In addition, the main advantage of these microtubule targeting agents is its less capability to have multidrug resistance.
Though majority of the clinically used drugs are tubulin inhibitors, they are added with several inherent limitations including the poor therapeutic window, lack of tumor specificity and low cytotoxicity. These limitations have led to the failure of potent tubulin inhibitors alone as an anticancer agent. Hence, the authors have made an attempt to find a suitable pharmacophoric model responsible for targeting microtubulin that may pave a way for the discovery of new generations of tubulin inhibitors with minimized limitations and greater potency. Among the different microtubule targeting agents, cryptophycin is a cyclic peptide having very good antitumor activity. Considering the above facts, our aim of the present work was to develop a quantitative pharmacophore model for cryptophycin to retrieve structurally diverse compounds by virtual screening with desired biological activity against cancer.
MATERIALS AND METHODS
The study was carried out in the Department of Pharmaceutical Technology, Drug Discovery Research Lab, University College of Engineering (BIT Campus), Anna University, Tiruchirappalli from February 2019 to July 2019.
Experimental Work: All the molecular modeling work were performed on a silicon graphics octane 2 R12000 computer with running lrix 6.5.12 and Catalyst HypoGen version 4.6 to generate pharmacophore models. All the chemical structures were built and minimized with the Catalyst and conformational analysis of each molecule was implemented using the pooling algorithm. HypoGen model was used for generating the pharmacophore of the training set of 20 compounds.
A pharmacophore is a representation of 3D arrangement of characteristic functional groups that are responsible for a desired biological activity. The best pharmacophore hypothesis is predicted based on the large difference between fixed cost and null cost, the total cost close to the fixed cost, the configuration cost less than 17 and the error cost which is dependent on the RMS (Root Mean Square) difference between the estimated and actual activities of the training set molecule. The generated pharmacophore hypothesis by using HypoGen model is shown in Table 1.
|
|||||||
| Hypothesis No. | Total Cost | Cost difference (Null cost - Total cost) | RMS deviation | Correlation | Features Definition | ||
| 1 | 86.432 | 57.714 | 0.774 | 0.948 | ADHR | ||
| 2 | 89.524 | 54.622 | 0.817 | 0.929 | ADHR | ||
| 3 | 90.201 | 53.945 | 0.913 | 0.910 | ADHR | ||
| 4 | 91.294 | 52.852 | 0.947 | 0.904 | AADH | ||
| 5 | 91.667 | 52.479 | 0.998 | 0.893 | AAHR | ||
| 6 | 92.012 | 52.134 | 1.020 | 0.888 | DHRR | ||
| 7 | 92.652 | 51.494 | 1.040 | 0.884 | AHRR | ||
| 8 | 95.241 | 48.905 | 1.008 | 0.903 | AAHR | ||
| 9 | 95.407 | 48.739 | 1.042 | 0.893 | AAHR | ||
| 10 | 96..051 | 48.095 | 1.179 | 0.852 | AAHR | ||
RMS deviation - Root-mean-square deviation, HBA - Hydrogen bond acceptor, HBD - Hydrogen bond donor, HA - Hydrophobic aliphatic, RA - Ring aromatic |
|||||||
Selection of Training set: A set of 45 cryptophycin inhibitors (IC50 value ranges from 0.022 nM to 1860 nM) were used as training set for the generation of pharmacophore hypothesis through HypoGen model. Structures of the generated 45 cryptophycin inhibitors are shown in Fig. 1. Training set of 20 compounds were selected from the database based on the quantitative predictive character such as structural diversity and biological activity. The maximum number of conformers generated was 250 and an energy range of 10 kcal/mol was chosen and the conformational analysis was implemented by using the pooling algorithm. All the structures were built and minimized with the Catalyst. The predicted IC50 values for estimating the activity against the human leukemia CCRF-CEM tumor cell lines were determined for the selected 20 training set molecules. The training set was further categorized into three, based on their experimental activity IC50 values, highly active (IC50≤50 nM), moderately active (50≤IC50≤500 nM), inactive (IC50>500 nM) to simplify the results.
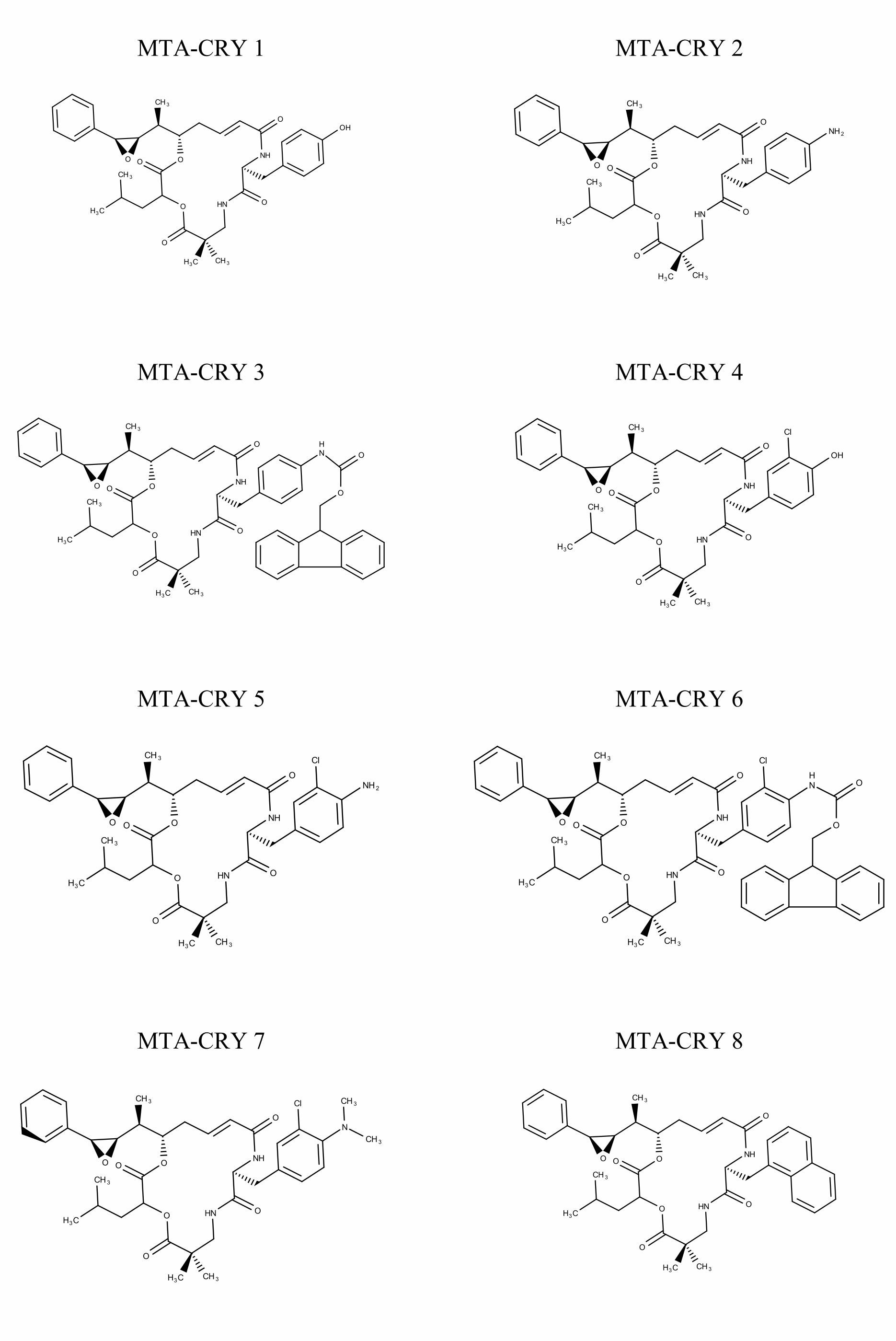
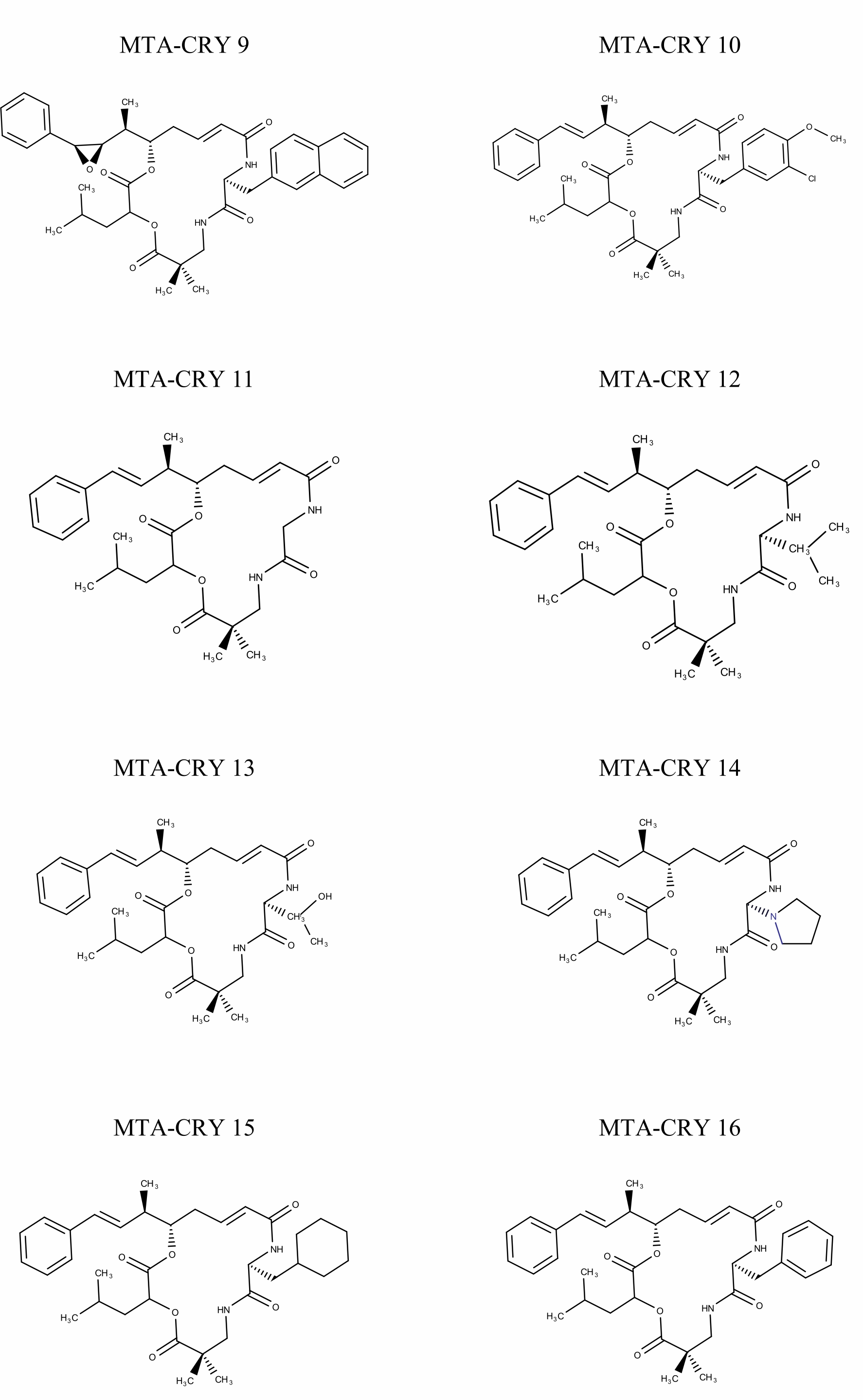
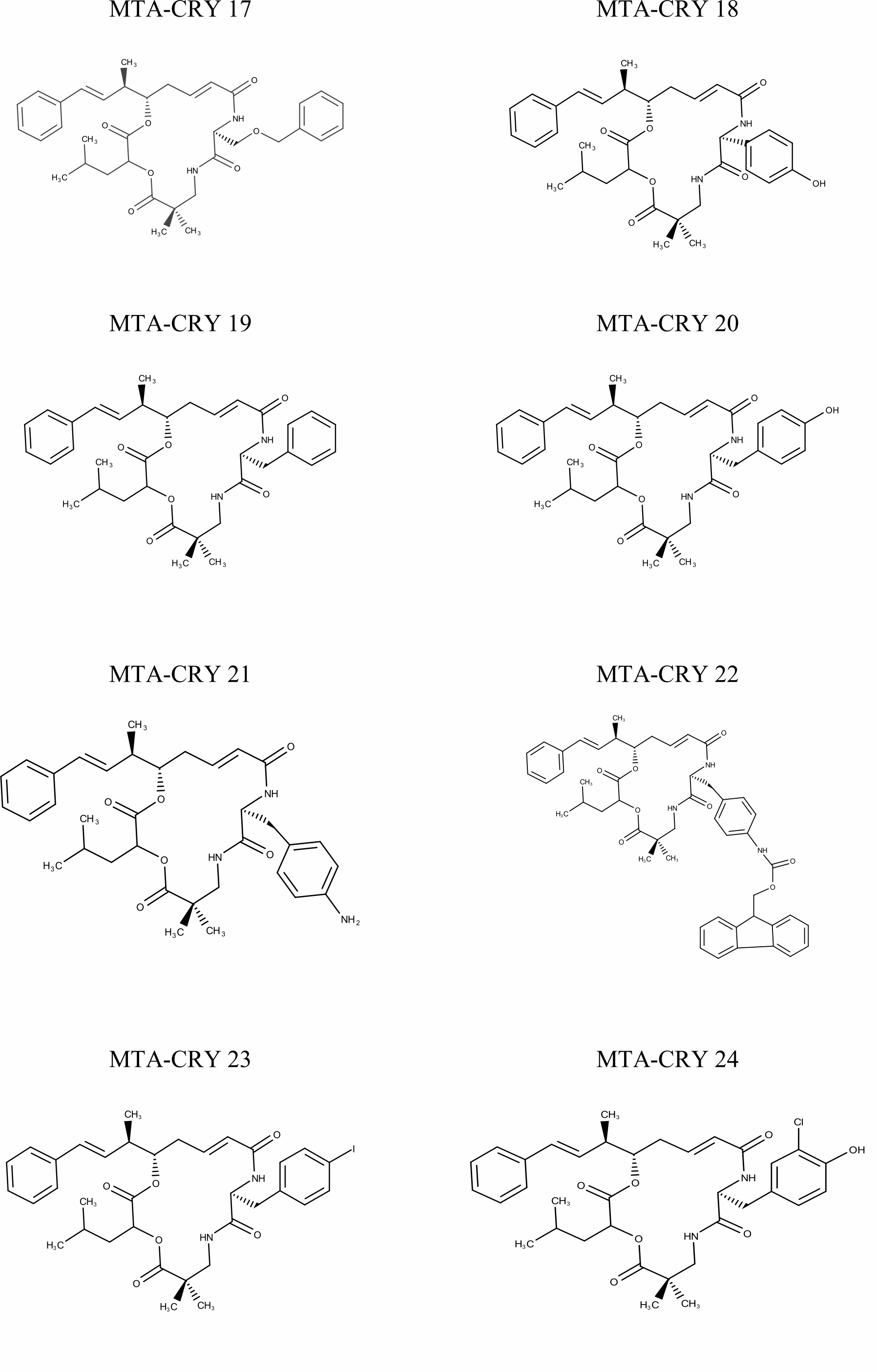
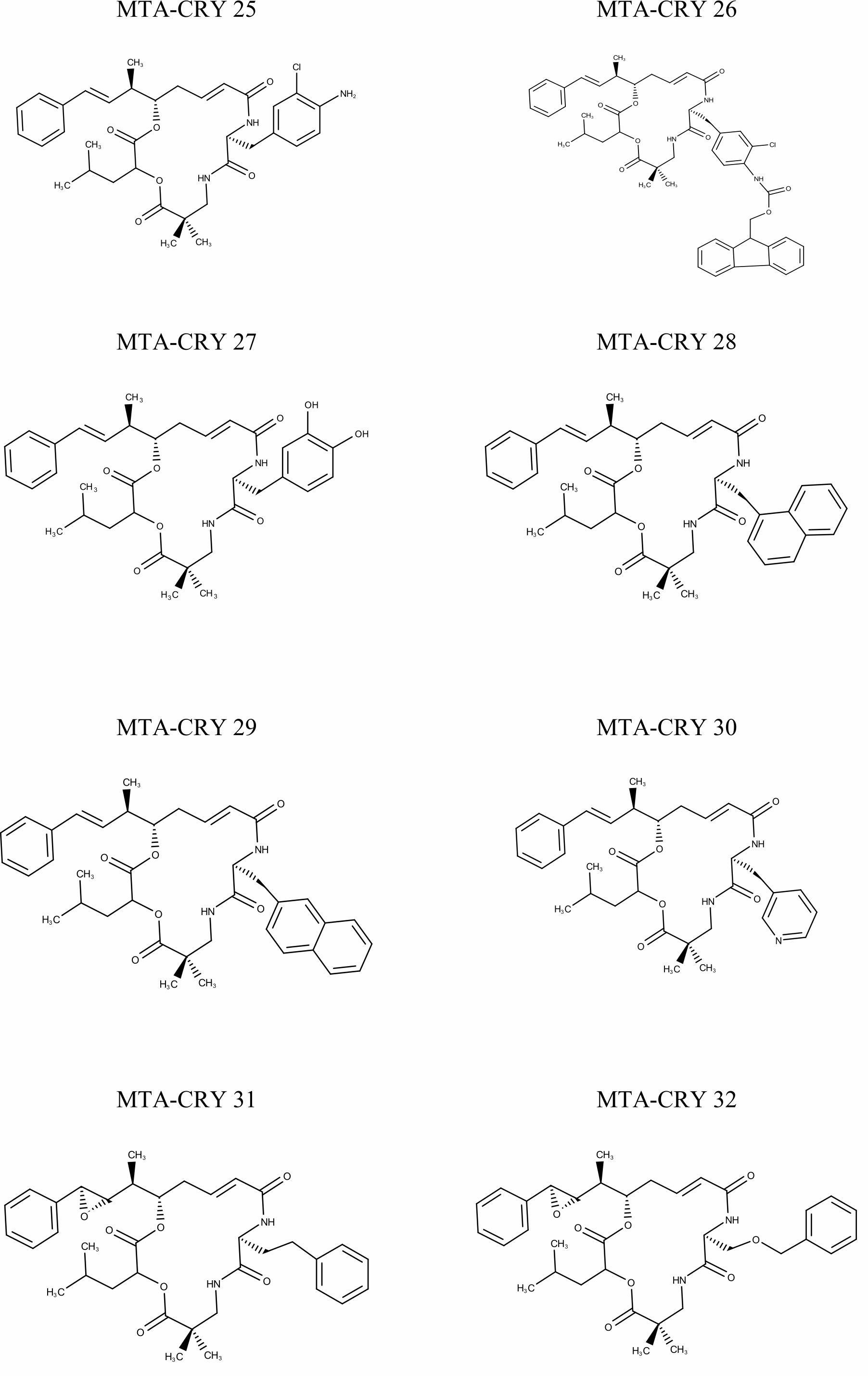
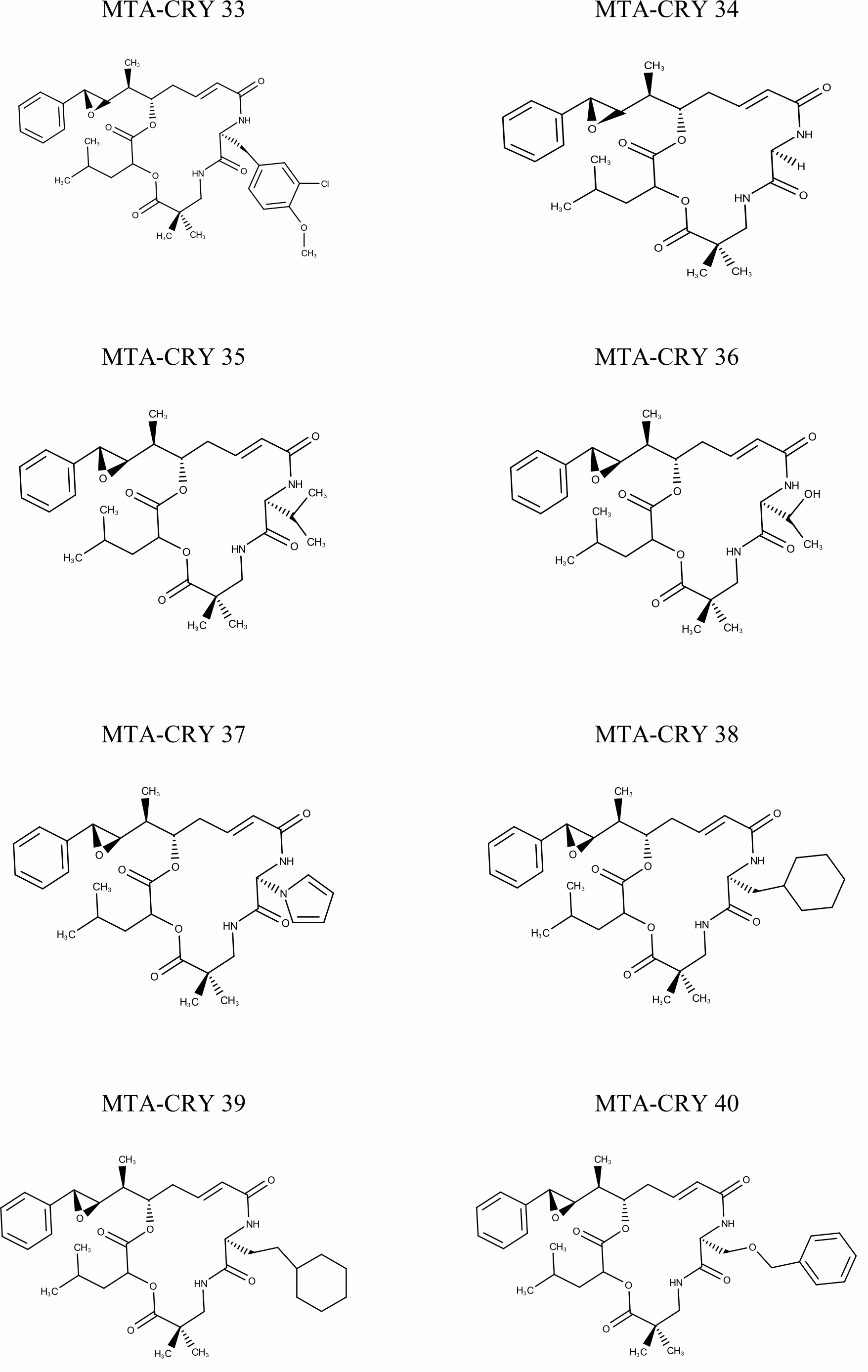
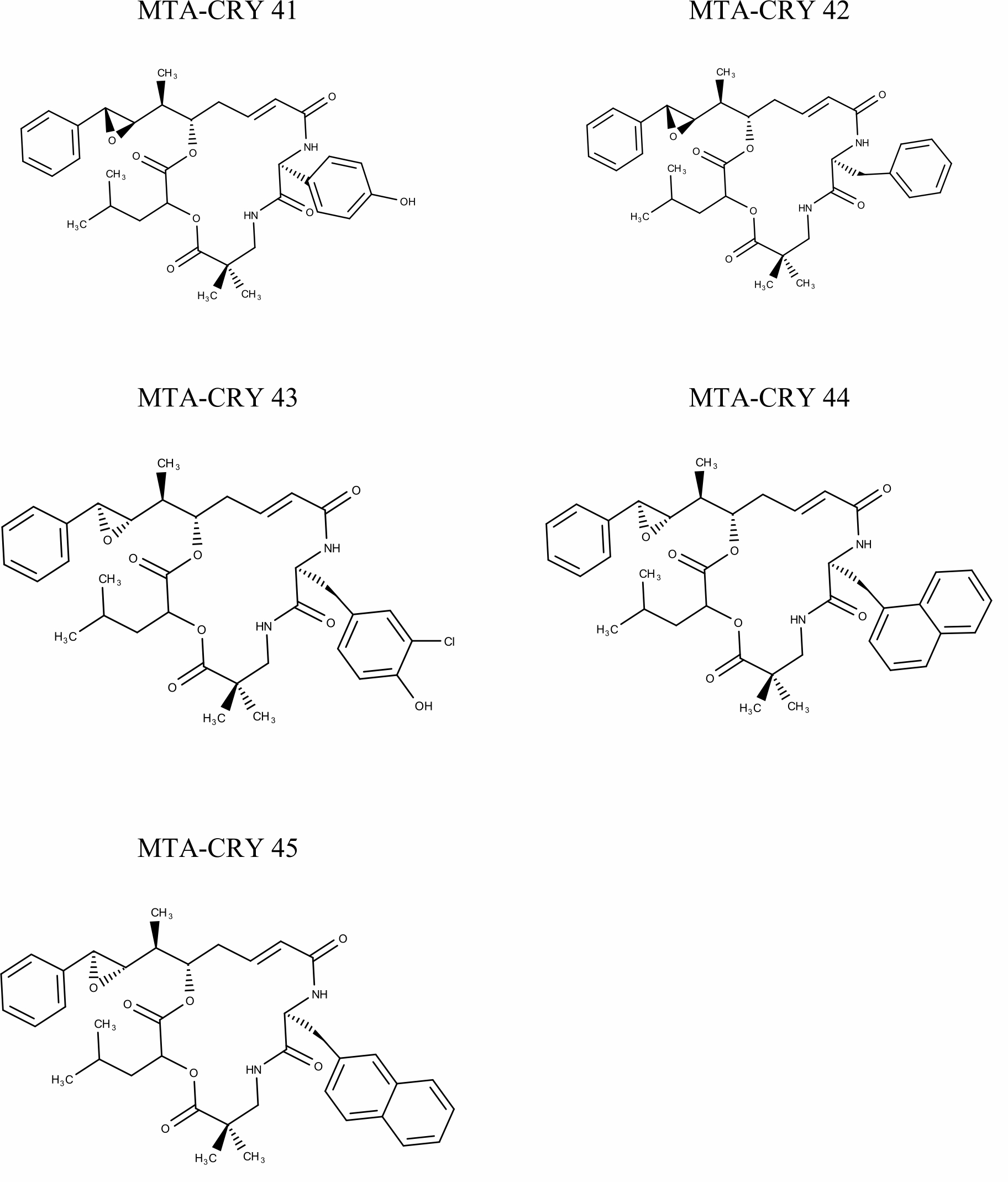 |
||||||||
Fig. 1 Pharmacophore Structures of Cryptophycin inhibitors |
|
MTA-CRY- Microtubule Targeted -Cryptophycin Derivatives |
Test Set: A test set of 18 compounds was selected from the database based on the quantitative predictive character such as structural diversity and biological activity. The maximum number of conformers generated was 250 and an energy range of 10 kcal/mol was chosen, and the conformational analysis was implemented by using the pooling algorithm. All the structures were built and minimized with the Catalyst. Hypothesis 1 was regressed against the selected 18 test set molecules to determine the predicted IC50 value for the pharmacological activity against human leukemia CCRF-CEM tumor cell lines. Similar to the training test, the test set was also further categorized into three, based on their experimental activity IC50 values, highly active (IC50≤50 nM), moderately active (50≤IC50≤500 nM), inactive (IC50>500 nM).
RESULTS
The known inhibitory activity data for the training set compounds was used to run the HypoGen protocol in the software. In our study, 10 different pharmacophore models were built based on the different cost value, total cost, RMS deviation and correlation for the training set compounds with experimental IC50 values for cryptophycin inhibitors and all were tested for four features. The result of the best pharmacophore model, Hypothesis 1 is displayed in Table 2. It was noticed that the predicted IC50 value was found to be 0.0226u 0.04 nM for compound MTA-CRY 33 indicating it is active whereas 18607d 2500 nM for compound MTA-CRY 14 indicated it is inactive. The results also revealed that of 20.6 compounds were predicted to be active (100%) accuracy, 7 as moderately active and the other 7 as inactive.
The best pharmacophore model, Hypothesis 1 was tailored from one hydrogen bond acceptor, one hydrogen bond donor, one hydrophobic aliphatic and one ring aromatic feature. Further, it is hypothesized that the best hypothesis should possess a greater difference between the fixed and null cost values. Hypothesis 1 in our results complies with the above said and had scored the values in the range from 86.432 to 96.051 for total cost, with cost difference in the range 48.095 to 57.714. The RMS deviation represents the quality of correlation between the predicted and actual activity data. Hypothesis in our study was observed to have 0.774 to 1.179 for RMS deviation and 0.852 to 0.948 for correlation. Among the 10 pharmacophore hypothesis, hypothesis No. 1 showed the total cost of 86.432 with cost difference of 57.714 and highest correlation value of 0.948 and 0.774 for RMS which exactly comply with the above said criterion.
|
||||||||
| S. No. | Compound No. | Actual IC50 | Predicted IC50 | Errora | Activity Scaleb | Estimated activity scaleb | ||
| 1 | MTA-CRY 2 | 10.11 | 12 | 1.2 | +++ | +++ | ||
| 2 | MTA-CRY 4 | 0.52 | 0.8 | 1.5 | +++ | +++ | ||
| 3 | MTA-CRY 7 | 0.054 | 0.04 | -1.3 | +++ | +++ | ||
| 4 | MTA-CRY 9 | 48 | 74 | 1.5 | +++ | ++ | ||
| 5 | MTA-CRY 14 | 1860 | 2500 | 1.4 | + | + | ||
| 6 | MTA-CRY 20 | 734 | 600 | -1.2 | ++ | ++ | ||
| 7 | MTA-CRY 22 | 1200 | 2200 | 1.8 | + | + | ||
| 8 | MTA-CRY 26 | 1250 | 260 | -4.8 | + | + | ||
| 9 | MTA-CRY 27 | 1301 | 2300 | 1.8 | + | + | ||
| 10 | MTA-CRY 28 | 1570 | 850 | -1.8 | + | + | ||
| 11 | MTA-CRY 30 | 997 | 930 | -1.1 | ++ | ++ | ||
| 12 | MTA-CRY 31 | 1620 | 1000 | -1.6 | + | + | ||
| 13 | MTA-CRY 33 | 0.022 | 0.14 | 1.5 | +++ | +++ | ||
| 14 | MTA-CRY 36 | 1060 | 260 | -4.1 | + | + | ||
| 15 | MTA-CRY 39 | 598 | 1000 | 1.7 | ++ | ++ | ||
| 16 | MTA-CRY 40 | 249 | 300 | 1.2 | ++ | ++ | ||
| 17 | MTA-CRY 41 | 274 | 280 | 1.0 | ++ | ++ | ||
| 18 | MTA-CRY 42 | 0.183 | 0.17 | -1.1 | +++ | +++ | ||
| 19 | MTA-CRY 44 | 327 | 270 | -1.2 | ++ | ++ | ||
| 20 | MTA-CRY 45 | 866 | 640 | -1.4 | ++ | ++ | ||
IC50 =Half maximal inhibitory concentration; a + = Predicted IC50 is higher than the experimental IC50; - = Predicted IC50 is lower than the experimental IC50; a value of 1 indicates that the estimated IC50 is equal to the experimental IC50, bActivity scale; IC50<50 nM = +++ (Highly active); IC50 50<500 nm = ++ (moderately active) and IC50>500 nM = + (Inactive) |
||||||||
The pharmacophore mapping, Hiphop protocol was used to map the 18 test set compounds on the generated pharmacophore hypothesis 1 for predicting the high fit test compound. The results of the biological activity against human leukemia CCRF-CEM tumor cell line and predicted IC50 value of the test set is shown in Table 3. Biological activity against the human leukemia CCRF-CEM tumor cell line, shows the predicted IC50 value for compound MTA-CRY 5 were found to be 0.69 nM indicating it as active whereas for the compound, (7a – 6b) 1600 nM for compound MTA-CRY 11 is MTA-CRY 11, it shows 1600 nM proving it to be inactive. Further, the result revealed that 7 active and 7 moderately active compounds were found to be with 100% accuracy for the predicted biological activity. On the other hand, out of 4 inactive compounds, 2 compounds depicted the predicted activity scale as active and the other two as inactive compounds. Hence this pharmacophore model hypothesis 1, can be utilized as a predictive tool for estimating biological activity of virtual compounds or for compounds designed on the basis of structure activity analysis.
|
||||||||
| S. No. | Compound No. | Actual IC50 | Predicted IC50 | Errora | Activity Scaleb | Estimated activity scaleb | ||
| 1 | MTA-CRY 1 | 4.1 | 2.6 | 0.6 | +++ | +++ | ||
| 2 | MTA-CRY 3 | 115 | 140 | 1.2 | ++ | ++ | ||
| 3 | MTA-CRY 5 | 0.58 | 0.69 | 1.1 | +++ | +++ | ||
| 4 | MTA-CRY 6 | 15.3 | 18.6 | 1.2 | +++ | +++ | ||
| 5 | MTA-CRY 8 | 35 | 42 | 1.1 | +++ | +++ | ||
| 6 | MTA-CRY 10 | 13.85 | 15.20 | 1.1 | +++ | +++ | ||
| 7 | MTA-CRY 11 | 2000 | 10 | -200.0 | + | +++ | ||
| 8 | MTA-CRY 16 | 1660 | 20 | -84.0 | + | +++ | ||
| 9 | MTA-CRY 17 | 856 | 750 | 0.8 | ++ | ++ | ||
| 10 | MTA-CRY 19 | 412 | 280 | -1.5 | ++ | ++ | ||
| 11 | MTA-CRY 23 | 607 | 430 | -1.4 | ++ | ++ | ||
| 12 | MTA-CRY 24 | 269 | 375 | 1.3 | ++ | ++ | ||
| 13 | MTA-CRY 25 | 500 | 400 | 0.8 | ++ | ++ | ||
| 14 | MTA-CRY 32 | 975 | 800 | 0.8 | ++ | ++ | ||
| 15 | MTA-CRY 35 | 1800 | 1600 | 0.8 | + | + | ||
| 16 | MTA-CRY 37 | 1350 | 1100 | 0.8 | + | + | ||
| 17 | MTA-CRY 38 | 8.8 | 12.6 | 1.4 | +++ | +++ | ||
| 18 | MTA-CRY 43 | 31 | 45 | 1.4 | +++ | +++ | ||
a + = Predicted IC50 is higher than the experimental IC50; - = Predicted IC50 is lower than the experimental IC50; a value of 1 indicates that the estimated IC50 is equal to the experimental IC50. bActivity scale; IC50<50 nM = +++ (Highly active); IC50 between 50 and 500 nm = ++ (moderately active) and IC50>500 nM = + (Inactive)
|
||||||||
DISCUSSION
Ligands most favorable 3D binding conformation with the receptor molecules are optimized based on docking studies by assumption that similar structural molecules can have similar biological activity than less similar or dissimilar molecules12. When receptor structural information is not known or unable to predict, Ligand Based Virtual Screening (LBVS) includes Similarity search, Pharmacophore-based virtual screening and Quantitative Structure. Activity Relationship are performed which plays an important role in drug discovery research. When numerous bioactive compounds are available, Pharmacophore Based Virtual Screening (PBVS) has been performed based on the ligands from the structural features of drug molecule. Pharmacophore models are generated based on the pharmacophoric features such as H-bond donors, H-bond acceptors, hydrophobic, aromatic and ionoizable groups and the 3-Dimensional space arrangements are responsible for compounds bioactivity13-18. In pharmacophore mapping, 2D drug structure activity information is transferred to 3D requirements for binding to the target biomolecule and then match these 3D properties with drug molecules from 3D database which predicts the chemical features of the drug molecules19-20.
Water miscibility and solubility of drug molecules significantly disturbs the route of administration and its pharmacokinetic parameters such as absorption, distribution, metabolism and elimination. Bioavailability is defined as the quantity of drug which reaches the systemic circulation. Factors like absorption, volume of distribution, half-life of the drug, first pass metabolism and rate of clearance affects the bioavailability. Solubility is the phenomenon of dissociation of the solute in a solvent to form a homogenous system, it has been considered as a major important parameter to attain the anticipated concentration of the drug in plasma and to achieve the desired pharmacological activity17.
Other factors such as poor aqueous solubility and slow dissolution rate can affect the bioavailability. In the present scenario about 40% to 70% of new drug molecules are considered to be highly hydrophobic compounds and intrinsically exhibit poor aqueous solubility21-22. Drug absorption and acceptable solubility in the intestinal part are the prerequisite in achieving desired concentration of drug in blood concentration and to produce a desired therapeutic effect23. Therefore, aqueous solubility has been considered as main criteria in identification of a lead molecule from discovery and development process17. In addition, solubility has been considered as a driving force in attaining the desired therapeutic effect besides important one for decision making and challengeable one with respect to the fate of the new drug molecule. Poor solubility is one of the major obstacles but desired solubility is the preferable one in the drug discovery and development process24. Medicinal chemist develops different strategies during optimization of lead compounds and to modify physicochemical properties to avoid the failure in the drug design and to save the time and investments. Last two decades in silico models (computational tools) such as Quantitative structure activity relationship and quantitative structure property relationship approach are adopted to identify the solubility in the physiological fluids like gastric and intestinal fluids based on physicochemical properties, 2D or 3D molecular structure and biology are considered by assuming that the similar chemical structure molecules have comparable properties with the combination of mathematical calculations. Stochastic artificial neural network and k-nearest neighbor methods are utilized to assess the similarity/dissimilarity of the drug molecule16,17,24-26.
Hydrogen bond/bonding (HB) and especially intramolecular HBs (IMHBs) are playing critical structure in synthetic molecular systems of drug designs, in molecular recognition as well as in the biological function27-28. In general, proteins and DNA are held together by hydrogen bonds. Whereas ubiquitous in nature of hydrogen bonds play important role in DNA base pairing, enzyme specificity, protein–ligand interactions and protein folding29. Drug molecule exhibits its therapeutic effect based on the interaction between the drugs and target proteins like ion channels, G-protein coupled receptors, kinase or nuclear hormone receptors, proteases, among others30. Ligand is stabilized at the interface of a protein structure based on the affinity between the ligand and protein complex through hydrogen bond by entropy gain or by transferring protein bound water molecules into the bulk solvent system29,31. Hydrogen bond is non-covalent interaction bond which is accountable for the 3D structure of biomolecule confirmation also affects the membrane transport and distribution within the biological systems 29. It is hypothesized that formation of IMHBs with optimal binding potency improves the membrane permeability and favorable intestinal absorption by polarity facilitating shield27. The cohesiveness of water liquid initiates the hydrophobic association in aqueous media is a significance of accommodation of hydrogen bond between water molecules which facilitates the drug solubility in both lipids and water32.
Some functional group in any molecule has the ability to accept or donate a hydrogen bond and contribute to water solubility which ultimately increases the hydrophilic nature of the drug molecules. On the contrary, the functional groups which do not form hydrogen bond bestow hydrophobic nature. Generally, there occurs an attraction between a minor positive charge of the hydrogenatom of a water molecule and the minor negative charge of the oxygen atom of drug molecule, forming a hydrogen bond. Due to this large electronegativity difference between them, it results in the formation of polar covalent bonds leading to strong dipole forces. Hence for the formation of hydrogen bond, the functional groups of the drug molecule necessarily have a dipole with electropositive hydrogen. This interaction is essential for the interaction among the drug and its biological target.
Subsequently, hydrophobicity of the drug molecule have important role in distribution, molecular recognition, protein folding, metabolism and excretion. In addition, transport and activity of drugs are predicted based on its polar and nonpolar phases of the solutes33.
Lastly, if the drug molecule has fewer aromatic rings, it has more probability of being developed whereas greater than three aromatic rings relates to poorer drugs with an increased risk in development. Aromatic ring (phenyl ring) in the drug molecular structure can improve its activity34.
In our study, the pharmacophore hypothesis 1 results predicted that one hydrogen bond acceptor, a hydrogen bond donor, one or two aromatic ring features and one hydrophobic feature as tubulin inhibitors which can best suits all the essential criterion for accurate drug targeting35-41.
Hence, this approach would definitely encourage scientists to focus on hydrogen bonding interactions, hydrophobic aliphatic interactions, aromatic effects for targeting microtubules, which have not yet been comprehensively analyzed.
CONCLUSION
The pharmacophore developed in this study for tubulin inhibitors showed distinct chemical features that may be responsible for its enhanced activity. The knowledge about the pharmacophore features is expected to be useful in identifying and designing inhibitors with greater selectivity towards tubulin. This work is intended to utilize the information to undertake 3D searches on large databases of drug like molecules to identify a new generation of tubulin inhibitors.
REFERENCES
- WHO., 2018. Cancer.
- WHO., 2021. Cancer-Key Facts.
- Hastings, K.G., D.B. Boothroyd, K. Kapphahn, J. Hu, D.H. Rehkopf, M.R. Cullen and L. Palaniappan, 2018. Socioeconomic differences in the epidemiologic transition from heart disease to cancer as the leading cause of death in the United States, 2003 to 2015. Ann. Internal Med., 169: 836-844
- Mitchison, T.J., 1988. Microtubule dynamics and kinetochore function in mitosis. Annual Rev. Cell Biol., 4: 527-545
- Jordan, M.A. and L. Wilson, 2004. Microtubules as a target for anticancer drugs. Nat. Rev. Cancer, 4: 253-265
- Lu, Y., J. Chen, M. Xiao, W. Li and D.D. Miller, 2012. An overview of tubulin inhibitors that interact with the colchicine binding site. Pharmaceut. Res., 29: 2943-2971.
- Saxton, W.M., D.L. Stemple, R.J. Leslie, E.D. Salmon, M. Zavortink and J.R. McIntosh, 1984. Tubulin dynamics in cultured mammalian cells. J. Cell Biol., 99: 2175-2186.
- Rusan, N.M., C.J. Fagerstrom, A.M.C. Yvon and P. Wadsworth, 2001. Cell cycle-dependent changes in microtubule dynamics in living cells expressing green fluorescent protein α-tubulin. Mol. Biol. Cell, 12: 971-980.
- Perez, E.A., 2009. Microtubule inhibitors: differentiating tubulin-inhibiting agents based on mechanisms of action, clinical activity, and resistance. Mol. Cancer Ther., 8: 2086-2095.
- Zhou, Q., A.K.K. Ching, W.K.C. Leung, C.Y.Y. Szeto and S.M. Ho, et al., 2011. Novel therapeutic potential in targeting microtubules by nanoparticle albumin-bound paclitaxel in hepatocellular carcinoma. Int. J. Oncol., 38: 721-731.
- Jordan, M.A., 2002. Mechanism of action of antitumor drugs that interact with microtubules and tubulin. Curr. Med. Chem. Anticancer Agent, 2: 1-17.
- Khanna, V. and S. Ranganathan, 2011. In silico approach to screen compounds active against parasitic nematodes of major socio-economic importance. BMC Bioinf.
- Leelananda, S.P. and S. Lindert, 2016. Computational methods in drug discovery. Beilstein J. Org. Chem., 12: 2694-2718.
- Adl, A., M. Zein and A.E. Hassanien, 2016. PQSAR: the membrane quantitative structure-activity relationships in cheminformatics. Expert Syst. with Appl., 54: 219-227.
- Prabu, S.L., R. Thirumurugan and T.N.K. Suriyaprakash, 2014. The role of the drug discovery, clinical, and regulatory affairs teams in turning a potent agent into a registered product. In: Reference Module in Chemistry, Molecular Sciences and Chemical Engineering, Reedijk, J. Elsevier, United States.
- Le, A.V., T.C.Q. Phan and T.H. Nguyen, 2015. In silico drug design: prospective for drug lead discovery. Int. J. Eng. Sci. Invent., 4: 60-70.
- Prabu, S.L., 2018. Drug discovery: current state and future prospects. In: Computer Applications in Drug Discovery and Development, Puratchikody, A., S.L. Prabu and A. Umamaheswari, IGI Global, United States.
- Kapetanovic, I.M., 2008. Computer-Aided Drug Discovery and Development (CADDD): In silico-chemico-biological approach. Chemico-Biol. Interact., 171: 165-176.
- Langer, T. and E.M. Krovat, 2003. Chemical features based pharmacophore and virtual library screening for discovery of new leads. Curr. Opin. Drug Discov. Devel., 6: 370-376.
- Lin, S.K., 2000. Pharmacophore perception, development and use in drug design. Molecules, 5: 987-989.
- Coltescu, A.R., M. Butnariu and I. Sarac, 2020. The importance of solubility for new drug molecules. Biomed. Pharmacol. J., 13: 577-583.
- Rane, S.S. and B.D. Anderson, 2008. What determines drug solubility in lipid vehicles: is it predictable? Adv. Drug Delivery Rev., 60: 638-656.
- Bergström, C.A.S. and P. Larsson, 2018. Computational prediction of drug solubility in water-based systems: qualitative and quantitative approaches used in the current drug discovery and development setting. Int. J. Pharmaceutics, 540: 185-193.
- Bergström, C.A.S., W.N. Charman and C.J.H. Porter, 2016. Computational prediction of formulation strategies for beyond-rule-of-5 compounds. Adv. Drug Delivery Rev., 101: 6-21.
- Schuhmacher, A., O. Gassmann and M. Hinder, 2016. Changing R&D models in research-based pharmaceutical companies. J. Transl. Med.
- Fuoco, D., 2015. Hypothesis for changing models: current pharmaceutical paradigms, trends and approaches in drug discovery. PeerJ PrePrints.
- Alex, A., D.S. Millan, M. Perez, F. Wakenhut and G.A. Whitlock, 2011. Intramolecular hydrogen bonding to improve membrane permeability and absorption in beyond rule of five chemical space. Med. Chem. Comm., 2: 669-674.
- Caron, G. and G. Ermondi, 2017. Why we need to implement intramolecular hydrogen-bonding considerations in drug discovery. Future Medic. Chem., 9: 1-5.
- Yunta, M.J.R., 2017. It is important to compute intramolecular hydrogen bonding in drug design? Am. J. Model. Optim., 5: 24-57.
- Prada-Gracia, D., S. Huerta-Yépez and L.M. Moreno-Vargas, 2016. Application of computational methods for anticancer drug discovery, design, and optimization. Bol. Med. Hosp. Infant. Mex., 73: 411-423.
- Testa, B., H.v. de Waterbeemd, G. Folkers and R. Guy 2007. Pharmacokinetic optimization in drug research: biological, physicochemical, and computational strategies. Wiley Online Library, United States.
- Kenny, P.W., C.A. Montanari, I.M. Prokopczyk, J.F.R. Ribeiro and G.R. Sartori, 2016. Hydrogen bond basicity prediction for medicinal chemistry design. J. Medic. Chem., 59: 4278-4288.
- Pietro, C. and F. Spyrakis, 2016. Hydrophobicity in Drug Design.
- Senese, S., Y.C. Lo, A.A. Gholkar, C.M. Li and Y. Huang et al., 2017. Microtubins: a novel class of small synthetic microtubule targeting drugs that inhibit cancer cell proliferation. Oncotarget, 8: 104007-104021.
- Kapetanovic, I.M., 2008. Computer-Aided Drug Discovery and Development (CADDD): In silico-chemico-biological approach. Chemico-Biol. Interact., 171: 165-176.
- Zavod, R.M. and J.J. Knittel, 2020. Drug design and relationship of functional groups to pharmacologic activity.
- Ritchie, T.J. and S.J.F. Macdonald, 2009. The impact of aromatic ring count on compound developability-are too many aromatic rings a liability in drug design? Drug Discov. Today, 14: 1011-1020.
- Zhou, Y., B. Di and M.M. Niu, 2019. Structure-based pharmacophore design and virtual screening for novel tubulin inhibitors with potential anticancer activity. Molecules.
- Niu, M.M., J.Y. Qin, C.P. Tian, X.F. Yan and F.G. Dong et al., 2014. Tubulin inhibitors: pharmacophore modeling, virtual screening and molecular docking. Acta Pharmacol. Sinica, 35: 967-979.
- Stanton, R.A., K.M. Gernert, J.H. Nettles and R. Aneja, 2011. Drugs that target dynamic microtubules: a new molecular perspective. Medic. Res. Rev., 31: 443-481.
- Zhao, W., J.K. Bai, H.M. Li, T. Chen and Y.J. Tang, 2015. Tubulin structure-based drug design for the development of novel 4 β-sulfur-substituted podophyllum tubulin inhibitors with anti-tumor activity. Sci. Rep.
How to Cite this paper?
APA-7 Style
Umamaheswari,
A., Prabu,
S.L., Puratchikody,
A. (2021). Microtubule Targeted Pharmacophore Study of Cryptophycin Derivatives for Effective Treatment of Cancer. Asian Journal of Emerging Research, 3(1), 25-31. https://doi.org/10.3923/ajerpk.2021.25.31
ACS Style
Umamaheswari,
A.; Prabu,
S.L.; Puratchikody,
A. Microtubule Targeted Pharmacophore Study of Cryptophycin Derivatives for Effective Treatment of Cancer. Asian J. Emerg. Res 2021, 3, 25-31. https://doi.org/10.3923/ajerpk.2021.25.31
AMA Style
Umamaheswari
A, Prabu
SL, Puratchikody
A. Microtubule Targeted Pharmacophore Study of Cryptophycin Derivatives for Effective Treatment of Cancer. Asian Journal of Emerging Research. 2021; 3(1): 25-31. https://doi.org/10.3923/ajerpk.2021.25.31
Chicago/Turabian Style
Umamaheswari, Appavoo, Sakthivel Lakshmana Prabu, and Ayarivan Puratchikody.
2021. "Microtubule Targeted Pharmacophore Study of Cryptophycin Derivatives for Effective Treatment of Cancer" Asian Journal of Emerging Research 3, no. 1: 25-31. https://doi.org/10.3923/ajerpk.2021.25.31

This work is licensed under a Creative Commons Attribution 4.0 International License.