
Phylogeny of Aedes Mosquitoes, Dengue and Chikungunya Viruses Along the Coastline of Kenya
-
Jonathan C. Ngala

1Kenyatta University, School of Medicine, Department of Medical Laboratory Science, P.O. Box 43844, Nairobi, Kenya
Muturi M. WanguiKenyatta University, School of Medicine, Department of Medical Laboratory Science, P.O. Box 43844, Nairobi, Kenya
Martin K. RonoPwani University Bioscience Research Centre (PUBReC), P.O. Box 195-80108, Kilifi, Kenya
| Received 28 Jul, 2020 |
Accepted 10 Oct, 2020 |
Published 15 Nov, 2020 |
Background and Objective: There are arthropod-borne disease outbreaks as a result of pathogen influx including arboviruses which are transmitted by strains of Aedes species that occur periodically in varying spots on the globe. This study aimed to determine the phylogenetic relationship of Aedes mosquitoes, Dengue and Chikungunya viruses along the Coastline of Kenya. Materials and Methods: This was necessitated by the paucity of documented information on phylogeny of the viruses and their vectors. A sampling of adult Aedes mosquitoes was done by the Bio-gent Sentinel trap and aspiration. Mosquitoes were identified and sorted based on their morphological features and molecular techniques. RNA was extracted using Trizole. Identification of Aedes species, Dengue and Chikungunya was done by a real-time polymerase chain reaction. Phylogeny trees were plotted by using the interactive tree of life. Results: Aedes aegypti was identified for the first time in this study along the Coastline of Kenya. Four serotypes of Dengue were identified, with Dengue-4 identified for the first time in this region. Only the East/Central/South African genotype of the Chikungunya virus was identified. Aedes mosquitoes, Dengue and Chikungunya had a close evolutionary relationship among themselves and to those from Africa, Asia and America. Conclusions: Aedes aegypti and Dengue virus serotype 4 were present along the Kenyan Coastline. There was a close evolutionary relationship among the identified haplotypes and to those from Africa, Asia and the American continent. Identification of new haplotypes indicates possible continuous evolution of the Aedes mosquito vectors and the viruses.
INTRODUCTION
Mosquitoes are responsible for the transmission of many pathogens, including viruses, bacteria and parasites between people, domestic and wild animals all over the world1. Phylogeny refers to the analysis and description of the evolutionary history of a group of organisms2. In biological studies, the primary goal of developing an evolutionary tree is to describe the evolutionary relationships between or among organisms; based on relative recency of common ancestry3. Phylogenetic analysis using analytical tools like parsimony phylogenetics could provide information on the common genetic changes (mutations) and modifications in biochemical pathways4. Such information could be crucial in connecting omics and the study of arboviral diseases. This could be by providing seamless, predictive, dynamic and multidimensional analysis. Such analysis could be utilized for timely diagnosis, detection and prognosis; identification of biomarkers and determination of effective treatment.
An increase in gene flow among Aedes aegypti populations between Africa and Saudi Arabia was previously reported; with phylogenetic relationships indicating that the two genetically distinct Aedes aegypti present in Saudi Arabia acquired their origin from the dual African ancestors5,6. In Africa, the phylogeny of West and East African isolates of Aedes aegypti s.l indicated the existence of two clades of origin5. The basal and derived/ upper clade: the basal clade had isolates mainly from West Africa while the upper clade had isolates mainly from East Africa. These findings demonstrated Aedes aegypti mosquitoes all over the world were composed of mosquitoes originating from one of the two matrilineages (basal and derived). Similarly, that there existed an evolutionary relationship among Aedes aegypti s.l populations from West and East Africa.
Earlier phylogenetic analysis of the Dengue virus demonstrated that DENV-1 isolates from Mombasa were related to those from Djibouti, China and Indonesia. DENV-2 isolates from Mombasa originated from those of West Africa: Senegal and Burkina Faso and were related to those from Asia. DENV-3 isolates from Mandera, Mombasa and Wajir originated from those of Brazil, Paraguay and Peru and were related to isolates from Pakistan, China and India7.
Phylogeny of the Chikungunya virus demonstrated that the Asian genotype emerged from the ECSA sometime between 1879 and 19278. More so, there were indications that the Asian genotype was related to strains from Asia and the Caribbean9. In Kenya, evolutionary studies on the ECSA genotype indicated that Kenyan isolates had a close evolutionary relationship among themselves and to those from China10. Therefore, this study was conducted to determine the phylogenetic relationship of Aedes mosquitoes, Dengue and Chikungunya viruses along the Coastline of Kenya. This was necessitated by the paucity of documented information on phylogeny of the viruses and their vectors.
MATERIALS AND METHODS
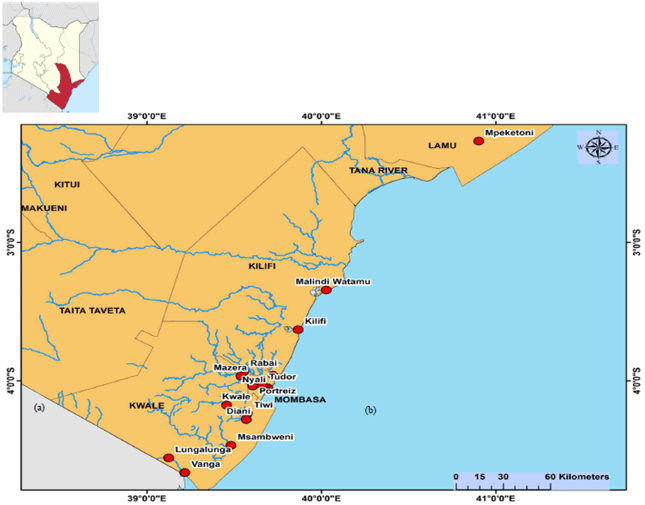
Study area: The study was carried out in selected sites along the coastline of Kenya from March 2018-December 2019 (Fig. 1). An indoor and outdoor sampling of adult Aedes mosquitoes were by Bio-gent sentinel (BG) traps (Biogents, Regensburg, Germany) and aspiration technique. Briefly, the BG traps were baited with solid carbon dioxide (dry ice) and suspended at least 0.5 m from the ground during outdoor sampling. Prokopack aspirators (John W. Hock, Gainesville, Florida, USA) were used for aspiration.
 |
||||
Fig. 1: Study sentinel sites, (a) A map of Kenya indicating the coastal region and (b) Sentinel study sites on the Coastline of Kenya |
Both BG trap and aspiration were performed twice a day; at dawn between 0500-1000 h and in the afternoon between 1500-1800 h. All captured mosquitoes were transported live in net cages to the field sorting insectary.
Morphological identification of Aedes mosquitoes: In the sorting insectary, the live Aedes mosquitoes were placed on a white filter paper in a Petri dish full of dry ice for 5 min. The stunning mosquitoes were identified morphologically to their species level, physiological status and sex before sorting. Morphological identification of mosquito species was by stereoscopic microscope (WITec, Germany)11 and according to manual “Mosquitoes of the Ethiopian Region”12.
RNA extraction and cDNA synthesis: Pools of Aedes mosquitoes (20 mosquitoes per pool) were homogenized by a mortar and pestle and RNA extracted using 1 mL of Trizol as earlier described5. Only the unfed and gravid mosquitoes were utilized in this analysis. The female blood-fed were excluded to avoid contamination of the virus which could be in the blood meal. cDNA synthesis was done on 10 μL of the extracted RNA to generate cDNA by using EcoDry Premix (Random hexamers) (Clontech Laboratories, Inc., Mountain View, CA, USA) in a procedure described earlier6,13.
Table 1: The primers for identification of Aedes mosquitoes |
||||
| Target | Primer name | Nucleotide sequences (5'-3') | Polarity | Product (bp) |
| Mosquito RNA | Act-2F | ATGGTCGGYATGGGNCAGAAGGACTC | Forwar | 683 |
| Act-8R | GATTCCATACCCAGGAAGGADGG | Revers | ||
| Ae. aegypti s.l | 18SFHIN | GTAAGCTTCCTTTGTACACACCGCCCGT | Forwar | 550 |
| Aeg.r1 | TAACGGACACCGTTCTAGGCCCT | Revers | ||
| Ae. tricholabis | UV | TGTGAACTGCAGGACACAT | Forwar | |
| Ae. pembaensis | PEM | GCATCGATGGGTTAATCATG | Revers | 405 |
| Ae. ocharaceous | OCH | CAAGCCGTTCGACCCTGATT | Revers | 501 |
| Ae. albicosta | ALB | Revers | ||
| Ae. fulgens | FUL | Revers | ||
| Ae. mcntoshi | MCN | Revers | ||
| Ae. fryeri | FRR | Revers | ||
| ND4 | ND4sb+ | TGATTGCCTAAGGCTCATGT | Forwar | 344 |
| ND4sb- | TTCGGCTTCCTAGTCGTTCAT | Revers | ||
Table 2: Primers and probes for identification of flaviviruses and alpha viruses |
||
| Primers and Probes | Nucleotide sequences | Tmo |
| Flavivirus | ||
| Flavi allS (Forward Primer) | 5'-TACAACATgATggggAARAgAgARAA-3' | 53.8 |
| Flavi all AS2 (Reverse Primer) | 5'-gTgTCCCAgCCNgCKgTgTCATCWgC-3' | |
| Flavi all AS4 (Reserve Primer) | 5'-gTgTCCCAGCCNgCKgTRTCRTC-3' | 80.4 |
| 3Pi (Probe) | FAM-Tg+gTWYATgT+ggYTNg+gRgC-NFQ-MGB | 50.3 |
| 3Pii (Probe) | FAM-CCgTgCCATATggTATATgTggCTgggAgC-NFQ-MGB | |
| 3Piii (Probe) | FAM-TTTCTggAATTTgAAgCCCTgggTTT-NFQ-MGB | |
| Pan-alphavirus | ||
| F2A (Forward Primer) | 5'- ATGATGAARTCIGGIATGTTYYT-3' | |
| R2A (Reverse Primer) | 5'-ATYTTIACTTCCATGTTCATCCA-3' | |
| R3A (Reverse Primer) | 5'-ATYTTIACTTCCATRTTCARCCA-3' | |
| R4A (Reverse Primer) | 5'-ATYTTIACTTCCATGTTGACCCA-3' | |
| ATTO425 (Probe) | -AT+GTT+GTC+GT+CIC+CIAT-BHQ1/LNA | |
Molecular Identification and sequencing of Aedes mosquitoes and Viruses: The mtNAD4 gene was utilized for the identification of Aedes mosquitoes using primers listed (Table 1) as earlier described5,6. Identification of serotypes of Dengue virus in the Aedes mosquitoes was based on the amplification of the target viral genes (E/NS1/NS5) in RNA using multiplex PCR with a panel of general flavivirus family primers (Table 2) as earlier described14. Samples tested positive for flavivirus were further tested with consensus primers for the Dengue virus. These primers were D1 and D2 and they target the E/NS1 junction of the virus genome. Only samples tested positive with Dengue consensus primers were further tested for the four Dengue serotypes using appropriate primers as described (Table 3)7,13. Identification of genotypes of the Chikungunya virus was based on the amplification of the target viral gene E1 in the RNA using multiplex PCR with the AgPath-ID One-step RT-PCR kit (Applied Biosystems, Carlsbad, Califonia, USA) using a similar method8,10 using a panel of general alphaviruses primers (Table 2). Samples tested positive for the alphavirus were further tested with conventional primers for the Chikungunya virus (Table 4).
Table 3: Primers for identification of serotypes of Dengue virus |
|||
| Primer | Gene/protein target | Primer sequence (5’-3’) | Position |
| FU1 | NS5 | TACAACATGATGGGAAAGAGAGAGAA | 9007-9032 |
| CFD3 | NS5 | GTGTCCCAGCCGGCGGTGTCATCAGC | 9308-9283 |
| D1 | E/NS1 | TCAATATGCTGAAACGCGCGAGAAACCG | 38-65 |
| D2 | E/NS1 | TTGCACCAACAGTCAATGTCTTCAGGTTC | 455-483 |
| TS1 | CGTCTCAGTGATCCGGGGG (D1 and TS1) | ||
| TS2 | CGCCACAAGGGCCATGAACAG (D1 and TS2) | ||
| TS3 | TAACATCATCATGAGACAGAGC (D1 and TS3) | ||
| TS4 | CTCTGTTGTCTTAAACAAGAGA (D1 and TS4) | ||
| D5-F | NS5 | TCAATATGCTGAAACGCGHGAG | 132-153 |
| D5-R | NS5 | GCGCCTTCNGNNGACATCCA | 764-783 |
Table 4: Primers for identification of Chikungunya virus |
|||
| Primer | Gene/protein target | Primer sequence (5’-3’) | Position |
| VIR 2052F | NSP4 | TGGCGCTATGATGAAATCTGGAATGTT | 6971-6997 |
| VIR 2052R | NSP4 | TACGATGTTGTCGTCGCCGATGAA | 7086–7109 |
| CHIKV-F | E1 | CGTGGTGTACAAAGGTGACG | 10524 |
| CHIKV-R | E1 | ACG CCG GGTAGTTGACTATG | 11170 |
The amplified gene products were cleaned from the gel by the MinElute PCR purification kit (Qiagen, Valencia, CA) and sequenced using Sanger high-throughput technique15.
Data analysis: Data were entered, cleaned and saved as comma-separated values (csv) extension in STATA 13 and transported to R. Proportions of Aedes species, sex orientation and their viral infection rates were determined. Generated nucleotide sequences were used for phylogenetic analysis. DNAbaser v.3.0 (http://www.dnabaser.com/articles/SNP) was used for editing bad calls in the raw chromatogram file generated from sequencing the forward and reverse strands. The deletion of the generated sequences of primers was done from the 5’ and 3’ ends. The sequences were subjected to the Basic Local Alignment Tool (BLAST) and GenBank database to compare them with available sequences and confirm the identity of the isolates. Formatting of the retrieved sequences compatible with alignment programs and identification of the correct reading frame for each sequence was done using the translation program at http://us.expasy.org/ tools/dna.html. The sequences were aligned for identification and removal of duplicate sequences using Clustal Omega v1.2.1, scored in T-coffee (http://tcoffee.crg.cat/) and viewed in jalview http://www.jalview.org/16. The sequences were manually adjusted in Se-Al software according to DNA sequence alignments for the preservation of codon homology17. Columns with more than one percent of gaps were removed from the alignment using trimAl v1.4.rev618. Using p-model test v1.4, Maximum likelihood trees were inferred using Randomised Accelerated Maximum Likelihood (RAxML) version 8.1.20 ran with model GTR-GAMMA-I for selecting the best- t model for the maximum likelihood analyses and plotting of phylogenetic trees using an interactive tree of life19. Branch supports were computed out of 1000 bootstrap replicates based on the Tamura Nei model20.
RESULTS
Phylogeny of Aedes mosquitoes along the coastline of Kenya: Aedes aegypti was identified for the first time in this study along the Coastline of Kenya. A total of 21 strains of Aedes species was identified.
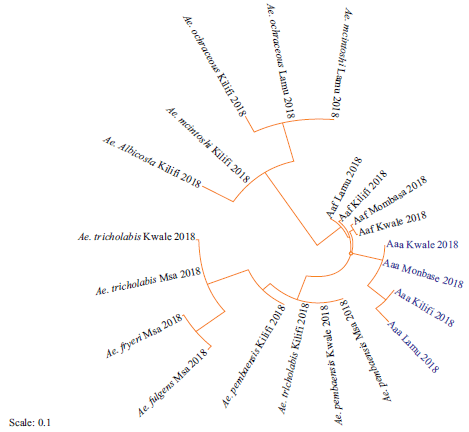 |
Fig. 2: Phylogeny of Aedes mosquitoes based on mtDNA NAD4 gene along the Coastline of Kenya Sequences aligned using clustal omega and the phylogenetic tree constructed using RAxML version 8.1.20. Newly identified strains are highlighted in blue and abbreviated with the site of collection and the year of isolation |
Four strains were identified for the first time in this study (highlighted in blue). Strains of Aedes aegypti s.l from Kilifi were closely related to those from Lamu. Similarly, strains from Kwale were closely related to those from Mombasa (Fig. 2). Phylogeny of the identified strains of Aedes mosquitoes along the Coastline of Kenya was compared with those previously identified in Kenya and other parts of the world (Fig. 3). Strains from West Africa (West Africa_KF939213) formed an out-group to the identified strains. All the new strains were in the in-group. All Aedes aegypti were in the same cluster and were closely related to strains from Mexico_JX297259, Uganda_DQ176838 and Brazil_AY906853. Aedes aegypti formosus strains from Lamu and Kilifi were closely related to EU446271.
Phylogeny of Dengue and chikungunya viruses along the coastline of Kenya: Four serotypes of Dengue virus were identified, that is, DENV-1, DENV-2, DENV-3 and DENV-4 serotypes as shown in Fig. 4. Sixteen new strains were identified with four identified for the first time (highlighted in blue). The phylogeny of these new strains was estimated as shown in Fig. 4: Strains from Kwale were closely related to those from Mombasa. Similarly, strains from Kilifi were closely related to those from Lamu. The phylogenetic relationship of the identified strains and those identified earlier in Kenya and all over the world was estimated (Fig. 5). DENV-1 strains were closely related to those previously isolated in Mombasa (KX812500), India (JN903581), France (DQ672556) and China (EF025110). Strains of DENV-2 were closely related to those previously reported in Mombasa (KX812508), Pakistan (KF041236) and China (KT187558).
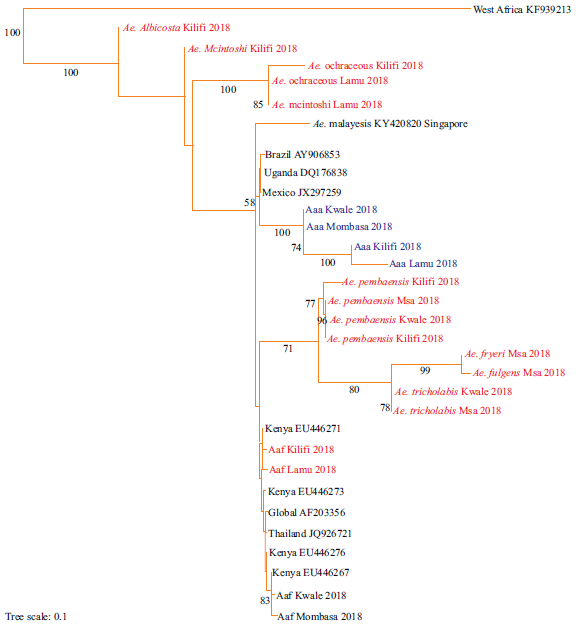 |
Fig. 3: Maximum likelihood tree of the mtDNA NAD4 of Aedes mosquitoes sampled along the Coastline of Kenya and other parts of the world Sequences aligned using clustal omega and the phylogenetic tree constructed using RAxML version 8.1.20. Bootstrap values for 1000 replicates are indicated at the major branch points. Newly identified strains are highlighted in red while strains of Aedes species identified for the first time are highlighted in blue |
New strains of DENV-3 were closely related to those previously isolated in Mombasa (KX812504; KX812511), Brazil (AY679147), India (AY770511) and China (KF954949). The strains of DENV-4 were closely related to those from Haiti (KP140942; JF262782).
In this study, only the ECSA genotype of the Chikungunya virus was identified (Fig. 6): Strains from Kwale and Mombasa formed a sister cluster. Similarly, strains from Kilifi formed a cluster with those from Lamu. Also, the phylogeny between newly identified strains and previous strains was estimated (Fig. 7). Identified strains were closely related to previous strains Mombasa_Chikv_HQ458254 and Lamu_Chikv_HQ456255. The ECSA strains were closely related to the Asian strains.
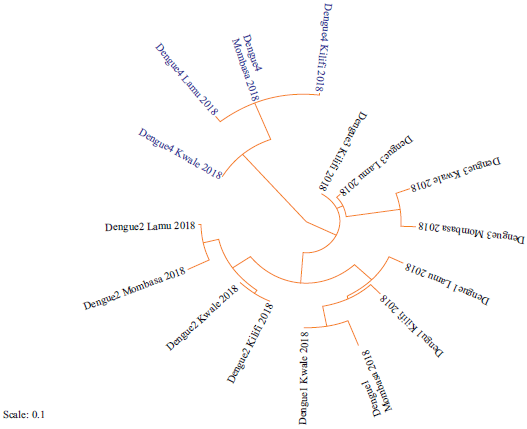 |
Fig. 4: Phylogeny of strains of Dengue serotypes based on the NSP5 coding regions along the Coastline of Kenya Sequences aligned using clustal omega and the phylogenetic tree constructed using RAxML version 8.1.20. Strains identified for the first time are highlighted in blue |
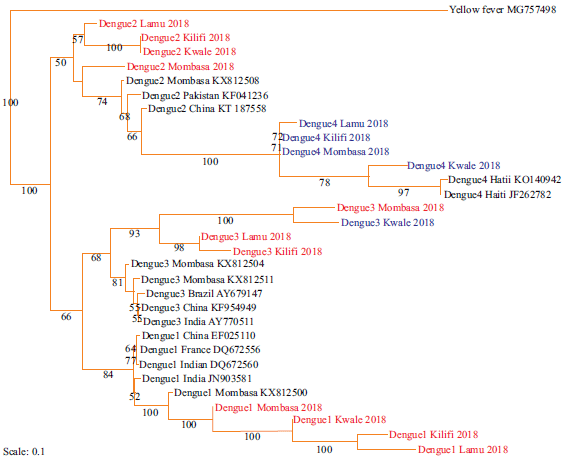 |
Fig. 5: Maximum likelihood tree of strains of Dengue serotypes based on the NSP5 coding regions along the Coastline of Kenya Sequences aligned using clustal omega and the phylogenetic tree constructed using RAxML version 8.1.20. Bootstrap values for 1000 replicates are indicated at the major branch points. New strains are highlighted in red. Strains identified for the first time are highlighted in blue |
 |
| Fig. 6: Phylogeny ofChikungunya strains based on nucleotide sequence of the partial E1 gene along the Coastline of Kenya. Sequences aligned using clustal omega and the phylogenetic tree constructed using RAxML version 8.1.20 |
 |
Fig. 7: Maximum likelihood phylogenetic tree among Chikungunya virus strains based on nucleotide sequence of the partial E1 gene along the Coastline of Kenya. Sequences aligned using clustal omega and the phylogenetic tree constructed using RAxML version 8.1.20. Bootstrap values for 100 replicates are indicated at the major branch points. New strains are highlighted in red |
DISCUSSION
The observation that strains of Aedes mosquitoes from West Africa formed an out-group implied they were related to members of in-group as they shared an orthologous gene group and could have given rise to the East African strains. Isolation of new strains in this study could be due to sampling of the entire Coastline for consecutive two years. More so, the evolution of these Aedes species may have occurred with time and space due to environmental selection pressure: challenges by insecticides, decrease in breeding sites and climate change especially increase in temperature.
The finding that both new and previously identified strains of Aedes mosquitoes from East Africa were in the in-group of the phylogenetic tree indicated a close evolutionary relationship among them and this conquers with the previous reports5. The observation that all strains from Kenya and Asian continent were in the in-group implied they originated from Aedes mosquitoes from West Africa and this may be attributed to the fact that there exist a lot of movement of goods and people between East Africa, West Africa and the Asian continent thus a possibility of trafficking these mosquitoes between these regions.
This study reported for the first time the presence of serotype DENV-4 in Aedes mosquitoes along the Coastline of Kenya. This may be due to the expansive geographical area of sampling and the fact that entomological assays were done contrary to earlier serological studies7,21. Identification of sixteen more new strains of Dengue could have been due to the expansion of the study area and duration of the study. The finding that all strains of Dengue virus from Kwale and Mombasa; and Kilifi and Lamu were forming distinct sister clades indicated a close phylogenetic relationship between them. This could be attributed to the proximity between Kwale and Mombasa and between Kilifi and Lamu with improved traffic between these respective Counties with travelers carrying strains of these viruses in their bodies or Aedes mosquitoes in automobiles. These viruses may end up in the same host cells resulting in the related progeny of recombinant strains thus their close relationship. All identified and previous strains of Dengue were in the in-group and related to those from the Asian continent. This may be because strains of the Dengue virus had a common ancestry therefore a higher possibility of similarity in their evolutionary path22. The presence of DENV-4 in Kenya could be due to the increased number of people traveling between China, India, Haiti and Kenya.
The identification of only one genotype of the Chikungunya virus was in line with reports from recent studies10,23. This, therefore, means the other genotypes of Chikungunya virus: West Africa and Asian genotypes have not yet been introduced in the region. The strains of Chikungunya shared an orthologous gene group meaning they originated from a common ancestor. Strains from Lamu were related to those from Kilifi; and those from Mombasa with those from Kwale; this may be due to proximity of these Counties and frequency of people traveling across borders of these regions.
Strains of Chikungunya were closely related to those from Asia (Thailand and Indonesia), suggesting a close phylogeny among them. In addition, the Asian genotypes originated from the East African genotype and this conquers with the previous reports8. This could be attributed to the historical relationships between African and Asian continents since the 17th and 18th centuries. The observation that Indian genotype formed an out-group could mean it gave rise to the West African, East, Central and Southern Africa genotypes.
CONCLUSION
Aedes aegypti was presented alongside the earlier identified Aedes species. Identification of new haplotypes indicates possible evolution of the Aedes mosquito vectors, Dengue and Chikungunya viruses along the Coastline of Kenya. There was a close phylogenetic relationship among the newly identified Aedes subspecies and haplotypes; and were related to those from West Africa, Asia and South America. This could be aided by enhanced traffic of goods and human beings among these parts of the continents carrying alongside with them Aedes mosquitoes infected with the viruses.
SIGNIFICANCE STATEMENT
This study discovers the existence of evolutionary relationships among Aedes mosquitoes, Dengue and Chikungunya virus from coastal Kenya and those from other parts of the world. The study also discovers the presence of Aedes aegypti and Dengue virus serotype-4 which had not been identified in the region. This knowledge will help researchers with information on the transmission patterns of the vectors and viruses and forecast future epidemics of Dengue and Chikungunya infections.
ACKNOWLEDGMENTS
The authors wish to thank all the residents of Kwale, Mombasa, Kilifi and Lamu Counties for their cooperation during field studies. This work was supported by the National Research Fund-Kenya, Kenya Medical Research Institute and Bernhard Nocht Institute for Tropical Medicine.
REFERENCES
- Kraemer, M.U., M.E. Sinka, K.A. Duda, A.Q. Mylne and F.M. Shearer et al., 2015. The global distribution of the arbovirus vectors Aedes aegypti and Ae. albopictus. eLIFE.
- Harrison, C.J. and J.A. Langdale, 2006. A step by step guide to phylogeny reconstruction. Plant J., 45: 561-572.
- Kitching, I.J., P.L. Forey, C.J. Humphries and D.M. Williams, 1998. Cladistics: The Theory and Practice of Parsimony Analysis. 2nd Edn., Oxford University Press, Oxford, UK.
- Abu‐Asab, M., M. Chaouchi and H. Amri, 2008. Evolutionary medicine: A meaningful connection between omics, disease and treatment. Proteomics-Clin. Applic., 2: 122-134.
- Moore, M., M. Sylla, L. Goss, M.W. Burugu and R. Sang et al., 2013. Dual African origins of global Aedes aegypti s. l. populations revealed by mitochondrial DNA. PLoS Negl. Trop. Dis.
- Al Ali, K.H., A.A. El-Badry, M. Al Ali, W.S.M. El-Sayed and H.A. El-Beshbishy, 2016. Phylogenetic analysis of aedes aegypti based on mitochondrial ND4 gene sequences in Almadinah, Saudi Arabia. Iran. J. Biotechnol., 14: 58-62.
- Konongoi, L., V. Ofula, A. Nyunja, S. Owaka and H. Koka et al., 2016. Detection of dengue virus serotypes 1, 2 and 3 in selected regions of Kenya: 2011-2014. Virol. J.
- Volk, S.M., R. Chen, K.A. Tsetsarkin, A.P. Adams and T.I. Garcia et al., 2010. Genome-scale phylogenetic analyses of chikungunya virus reveal independent emergences of recent epidemics and various evolutionary rates. J. Virol., 84: 6497-6504.
- Yoon, I.K., M.T. Alera, C.B. Lago, I.A. Tac-An and D. Villa et al., 2015. High rate of subclinical chikungunya virus infection and association of neutralizing antibody with protection in a prospective cohort in the Philippines. PLoS Negl. Trop. Dis.
- Konongoi, S.L., A. Nyunja, V. Ofula, S. Owaka and H. Koka et al., 2018. Human and entomologic investigations of chikungunya outbreak in Mandera, Northeastern Kenya, 2016. PloS One.
- Gillies, M.T. and B. De Meillon, 1968. The Anophelinae of Africa south of the Sahara (Ethiopian Zoogeographical Region). 2nd Edn., South Africa Institute for Medical Research, Johannesburg, South Africa.
- Huang, Y.M., 2002. A pictorial key to the mosquito genera of the world, including subgenera of Aedes and Ochlerotatus (Diptera: Culicidae). Insecta Koreana, 19: 1-130.
- Lanciotti, R.S., C.H. Calisher, D.J. Gubler, G.J. Chang and A.V. Vorndam, 1992. Rapid detection and typing of dengue viruses from clinical samples by using reverse transcriptase-polymerase chain reaction. J. Clin. Microbiol., 30: 545-551.
- Kuno, G., G.J.J. Chang, K.R. Tsuchiya, N. Karabatsos and C.B. Cropp, 1998. Phylogeny of the genus Flavivirus. J. Virol., 72: 73-83.
- Sanger, F., S. Nicklen and A.R. Coulson, 1977. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. USA., 74: 5463-5467.
- Sievers, F. and D.G. Higgins, 2014. Clustal omega. Curr. Protocols Bioinform., 48: 3.13.1-3.13.16.
- Rambaut, A., 1996. Se-Al: Sequence alignment editor.
- Capella-Gutiérrez, S., J.M. Silla-Martínez and T. Gabaldón, 2009. TrimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics, 25: 1972-1973.
- Stamatakis, A., 2014. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics, 30: 1312-1313.
- Tamura, K. and M. Nei, 1993. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol., 10: 512-526.
- Ngoi, C.N., M.A. Price, B. Fields, J. Bonventure and C. Ochieng et al., 2016. Dengue and chikungunya virus infections among young febrile adults evaluated for acute HIV-1 infection in coastal Kenya. PLoS One.
- Añez, G., D.A. Heisey, L.M. Espina, S.L. Stramer and M. Rios, 2012. Phylogenetic analysis of dengue virus types 1 and 4 circulating in Puerto Rico and Key West, Florida, during 2010 epidemics. Am. J. Trop. Med. Hyg., 87: 548-553.
- Sergon, K., C. Njuguna, R. Kalani, V. Ofula and C. Onyango et al., 2008. Seroprevalence of chikungunya virus (CHIKV) infection on Lamu Island, Kenya, October 2004. Am. J. Trop. Med. Hyg., 78: 333-337.
How to Cite this paper?
APA-7 Style
C. Ngala,
J., Wangui,
M., K. Rono,
M. (2020). Phylogeny of Aedes Mosquitoes, Dengue and Chikungunya Viruses Along the Coastline of Kenya. Asian Journal of Emerging Research, 2(4), 200-211. https://doi.org/10.3923/AJERPK.2020.200.211
ACS Style
C. Ngala,
J.; Wangui,
M.; K. Rono,
M. Phylogeny of Aedes Mosquitoes, Dengue and Chikungunya Viruses Along the Coastline of Kenya. Asian J. Emerg. Res 2020, 2, 200-211. https://doi.org/10.3923/AJERPK.2020.200.211
AMA Style
C. Ngala
J, Wangui
M, K. Rono
M. Phylogeny of Aedes Mosquitoes, Dengue and Chikungunya Viruses Along the Coastline of Kenya. Asian Journal of Emerging Research. 2020; 2(4): 200-211. https://doi.org/10.3923/AJERPK.2020.200.211
Chicago/Turabian Style
C. Ngala, Jonathan, Muturi M. Wangui, and Martin K. Rono.
2020. "Phylogeny of Aedes Mosquitoes, Dengue and Chikungunya Viruses Along the Coastline of Kenya" Asian Journal of Emerging Research 2, no. 4: 200-211. https://doi.org/10.3923/AJERPK.2020.200.211

This work is licensed under a Creative Commons Attribution 4.0 International License.
Division of Scientific Publishing
ACE College for Women
Faisalabad-38090, Pakistan
ISSN: 2663-4988 / 2664-5211
This work is licensed under a Creative Commons Attribution 4.0 International License.